
Фактор некроза опухоли (ФНО): многофункциональный воспалительный цитокин
Фактор некроза опухоли (ФНО, TNF-α) – это внеклеточный белок, относится к воспалительным цитокинам. Его в основном вырабатывают макрофаги и моноциты. Свое название получил из-за того, что стимулирует некроз некоторых раковых опухолей.
Статья основана на выводах 239 научных исследований
В статье цитируются такие авторы, как:
Обратите внимание, что буквы в скобках (1, 2, 3 и т.д.) являются кликабельными ссылками на рецензируемые научные исследования. Вы можете перейти по этим ссылкам и ознакомиться с первоисточником информации для статьи.
Тремя наиболее распространенными воспалительными цитокинами, ответственными за хронические воспалительные заболевания, являются фактор некроза опухоли (ФНО), интерлейкин-1beta (IL-1b) и интерлейкин-6 (IL-6).
ФНО играет важную роль во многих заболеваниях, включая аутоиммунные болезни, сердечно-сосудистые заболевания и рак. ФНО повышает уровень С-реактивного белка, который является одним из основных показателей для оценки воспаления. Однако, С-реактивный белок недостаточно чувствителен для выявления более низких, но хронических уровней фактора некроза опухоли.
ФНО повышает уровень С-реактивного белка, который является одним из основных показателей для оценки воспаления. Однако, С-реактивный белок недостаточно чувствителен для выявления более низких, но хронических уровней фактора некроза опухоли.
Сегодня науке известны 18 цитокинов, которые вызывают апоптоз клеток. Все они включены в семейство факторов некроза опухоли. Но первыми среди этих цитокинов были обнаружены фактор некроза опухоли (ФНО-альфа) и лимфотоксин-альфа (ранее назывался ФНО-бетта).
Содержание статьи
Подробнее о факторе некроза опухоли
Ядерный фактор транскрипции (NF-kappaB) также широко участвует в формировании воспаления. Все эти четыре белка (ФНО, IL-6, IL-1b и NF-kappaB) участвуют в процессе, который мы называем хроническим воспалением.
ФНО может быть увеличен как при состоянии доминирования иммунных клеток типа Th2, так и при доминировании клеток типа Th3. Важно понимать, что вы можете иметь повышенный уровень ФНО локально в таких областях, как гипоталамус головного мозга или в кишечнике, и это не будет показано в анализах крови.
Многие виды клеток высвобождают ФНО, а затем он продолжает стимулировать другие иммунные клетки. ФНО в основном высвобождается макрофагами и моноцитами, но его также производят дендритные клетки, иммунные Т-клетки, жировые клетки и фибробласты.
Затем фактор некроза опухоли воздействует на различные клетки. В частности, он сильно влияет на эндотелиальные клетки, которые выстилают наши кровеносные сосуды, что вызывает проблемы с кровеносными сосудами и это сильно стимулирует злокачественные опухоли, так как вызывает ангиогенез и повышенное образование кровеносных сосудов (гиперваскуляризацию). ФНО также может вызвать болезни сердца (атеросклероз), почек и проблемы с работой головного мозга.
ФНО также отрицательно влияет на клетки кишечника, вызывая гибель этих клеток, нарушение “герметичности” кишечника, формируя воспалительные заболевания кишечника.
Фактор некроза опухоли стимулирует макрофаги и эффекторные Т-клетки, что приводит к увеличению продукции воспалительных цитокинов и устойчивости к апоптозу (что способствует развитию рака).
Положительная роль фактора некроза опухоли
ФНО индуцирует сон и увеличивает длительность сна без его быстрой части. (1)
Поэтому, когда мы хотим заснуть, было бы хорошо, чтобы ФНО был немного выше в ночное время. Фактор некроза опухоли повышается естественным образом у здоровых людей в ночное время.
Продолжительность дневного сна при болезни Альцгеймера связана со степенью функционального нарушения головного мозга у больного, что является результатом повышенного воспаления. (2)
Другими примерами дневной сонливости при хронических воспалительных состояниях являются болезнь Паркинсона, черепно-мозговая травма, инсульт, сердечная недостаточность и диабет 2-го типа. (2)
ФНО – это прямой разрушитель жира, который приводит к инсулинорезистентности в жировых клетках (3), и к инсулинорезистентности в мышечных клетках. (4) Очень часто люди с повышенным хроническим воспалением выглядят худыми..png) Высокий ФНО заставляет вас есть меньше и подавляет получение глюкозы жировыми клетками.
Высокий ФНО заставляет вас есть меньше и подавляет получение глюкозы жировыми клетками.
Фактор некроза опухоли подавляет аппетит, снижая уровень орексина. (5, 6, 2)
Если вы уменьшите ФНО, то станете более голодными и ваше тело будет хранить больше жира. Поэтому неудивительно, что терапия препаратами анти-ФНО приводит к увеличению веса – в среднем на 5,5 кг всего за 12 недель. (7)
Влияние цитокинов, в том числе фактора некроза опухоли (ФНО), на органы при уремии (источник)Низкие дозы ФНО, вводимые мышам, вызывали резистентность гипоталамуса к инсулину и лептину, что способствовало ожирению. (8)
Даже относительно низкий уровень ФНО снижает синтез альдостерона в ответ на выработку АКТГ (предшественник кортизола), который повышается в ответ на стресс и интенсивную физическую нагрузку. Это снижает кровяное давление.
Физические упражнения повышают АКТГ (адренокортикотропный гормон), который обычно помогает нашему организму сохранять соль, увеличивая альдостерон.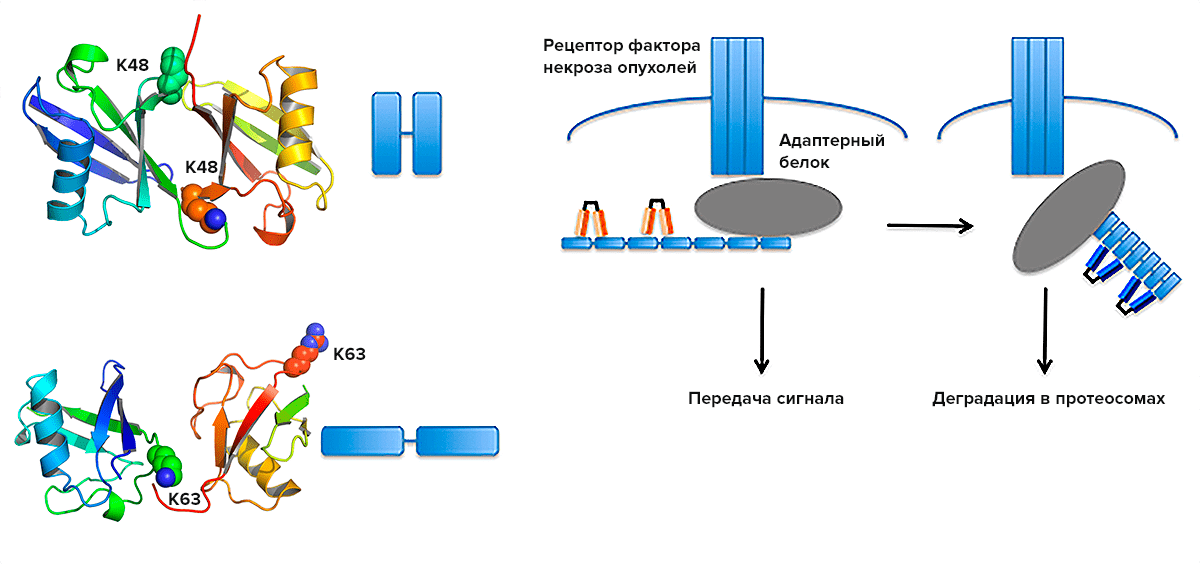 Однако если у вас есть воспаление, то выработка альдостерона будет ограничена, и если мы много потеем, это может вызвать низкий уровень соли в организме.
Однако если у вас есть воспаление, то выработка альдостерона будет ограничена, и если мы много потеем, это может вызвать низкий уровень соли в организме.
Негативная роль фактора некроза опухоли
Люди, которые имеют повышенный уровень воспаления, и поэтому проявляющие усталость и апатию, часто прибегают к различным психологическим трюкам или к изменению своего мышления. Но эти шаги только усилят ваш невротизм.
Если у вас повышен уровень ФНО и вы снижаете его, это положительно скажется на настроении и мотивации. Фактор некроза опухоли играет важную роль во многих заболеваниях и является цитокином, который является убийцей вашей мотивации и производительности труда.
ФНО также может нанести длительный вред, повреждая ваши митохондрии. (10)
ФНО заставляет вас уставать, снижает настроение и уменьшает мозговую и и физическую работоспособность, подавляя орексин. (5, 6, 2) Орексин – очень важный нейромедиатор для многих функций организма.
(5, 6, 2) Орексин – очень важный нейромедиатор для многих функций организма.
Орексин выполняет ряд ключевых ролей в получении памяти на недавние события, а также в укреплении долговременной памяти. (2) Следовательно, если у вас есть повышенное воспаление, то это повредит правильной работе мозга.
Фактор некроза опухоли также может снижать уровень гормонов щитовидной железы, вызывая “синдром низкого уровня Т3”. (11) Кроме того, ФНО способен снизить уровень тестостерона (немного, но не значительно). (12)
Фактор некроза опухоли (ФНО) участвует в развитии сепсиса (источник)ФНО замедляет заживление ран, а это значит, что вам потребуется больше времени для восстановления после физических упражнений или травм. (13, 14)
Фактор некроза опухоли способен формировать синдром – “дырявый кишечник”, который приводит к развитию некоторых воспалительных заболеваний кишечника. (15)
Хронически повышенный уровень ФНО также может нарушать циркадный ритм и вызывать утомляемость в течение дня. (2)
ФНО может не дать вашему телу перейти в состояние кетоза. (16)
Заболевания с повышенным уровнем ФНО
- Аутоиммунные заболевания: в целом (18), рассеянный склероз (19), болезнь Бехчета (20), волчанка (21), склеродермия (22), саркоидоз (22), гнойный гидраденит (23), анкилозирующий спондилит (23), узловатая эритема. (24)
- Болезни сердца: в целом (25), сердечная недостаточность (26), атеросклероз (26), инсульт (27)
- Рак в целом (28), меланома (18)
- Инсулинорезистентность (29)
- Сахарный диабет 2-го типа (30) и диабетическая невропатия (52)
- Болезнь Альцгеймера (31)
- Болезнь Паркинсона (22)
- Боковой амиотрофический синдром (32)
- Депрессия (33). У людей с депрессией уровень ФНО примерно на 3,97 пг/мл выше, чем у здоровых людей. (34)
- Воспалительные заболевания кишечника (35, 36), болезнь Крона / язвенный колит. (37)
- Ревматоидный артрит (23)
- Остеопороз (26)
- Псориаз (23)
- Экзема (22)
- Астма (22)
- ХОБЛ (38) ФНО стимулирует различные молекулы, которые рекрутируют эозинофилы, нейтрофилы и лимфоциты в дыхательные пути. ФНО может индуцировать резистентность к применяемым кортикостероидам. (39)
- Шизофрения (33)
- Биполярное расстройство (41, 42)
- Анорексия
- Синдром поликистозных яичников (СПКЯ). (44) ФНО сдвигает выработку кортизола в тестостерон, тем самым вызывая развитие СПКЯ. (45)
- Синдром хронической усталости (46)
- Фибромиалгия (47, 48)
- Хронические заболевания печени (53), включая жировую болезнь печени (22)
- Другие: шум в ушах (49), эпилепсия (22), муковисцидоз (50), вызванная химиотерапией невропатия (51)
Наиболее важные факторы, влияющие на ФНО
Двумя основными факторами, вызывающими хронический повышенный уровень ФНО, являются лектины (54) и ожирение (55), поскольку жировые клетки продуцируют ФНО. Если бы мы ели меньше, то у многих людей исчезли бы проблемы с воспалением.
Другими вероятными причинами являются хроническое недосыпание (56) и высокий уровень глюкозы в крови (57) из-за переедания, особенно продуктов с высоким гликемическим индексом.
Еще одними распространенными факторами являются недостаток физической активности (58), избыток алкоголя (59), курение (60) и недостаточное количество фруктов и овощей в питании. (61)
Ультра-фиолетовое излучение (солнечные лучи) сначала резко повышает уровень ФНО (62, 63, 64), но в конечном итоге снижает его. Вероятно, именно по этой причине некоторые люди испытывают усталость после солнечных ванн.
Недавние исследования показывают, что фактор некроза опухоли потенциально подавляется физической нагрузкой (в частности, миокинами или белками, продуцируемыми мышцами, такими как IL-6, но с противовоспалительным свойством). (65) Однако слишком большие физические нагрузки или марафоны повышают уровень ФНО. (66)
Физическая активность снижает воспаление, в том числе фактор некроза опухоли (ФНО) (источник)Инфекция также может повышать уровень ФНО. Бактериальные, но не вирусные, инфекции более вероятны, если у вас повышен уровень ФНО. (67)
Другие стимулы повышения уровня фактора некроза опухоли
- Окислительный стресс – активность ФНО зависит от генерации АФК (активных форм кислорода). (68)
- Лектины – для людей, чувствительных к лектину (возможно, четверть населения)
- Солнечные лучи – как UVA, так и UVB, резко повышают ФНО. (62, 63, 64). Но пребывание под солнечными лучами очень полезно для здоровья.
- Недостаток сна (56)
- Хроническая бессонница или сон в дневное время. (69)
- Высокий уровень глюкозы (57), обычно из-за продуктов питания с высоким гликемическим индексом. (70)
- Недостаток физической нагрузки (58)
- Недостаток стойкого крахмала в питании (71)
- Нарушение циркадного ритма (72)
- Избыток приема алкоголя (59)
- Курение (60)
- Диета с высоким содержанием жиров (особенно с пальмитиновой кислотой (73), но это не означает, что нужно переходить на диету с низким содержанием жиров. (74)
- Недостаток фруктов и овощей, которые содержат фитохимические вещества, подавляющие ФНО и другие молекулы воспаления. (61)
- Тяжелые металлы (75)
- Некоторые токсины (76)
- Дефицит магния (77)
- Дефицит цинка (78)
- Дефицит витамина D (79, 80, 81)
- Дефицит хрома (82)
- Дефицит холина (83) – у людей, потреблявших более 360 мг в день, концентрация ФНО была на ниже 12%, чем у тех, кто потреблял холина меньше.
- Кофе – у тех людей, кто пьет кофе, в среднем на 28% выше уровень ФНО, но, возможно, люди с высоким уровнем ФНО имеют усталость, поэтому они чаще пьют кофе. Вам стоит проверить свои показатели ФНО когда вы пьете кофе, и когда его не употребляете.
- Растения: ягоды годжи (84), алоэ (только в раковых клетках) (85), гриб Рейши (86) – снижает, если ФНО высокий (87), Андрографис (85)
- Аписмин (содержится в меде и маточном молочке) (88).
- Арабиногалактан (88)
- Вазопрессин (89)
- IGF-1 (соматомедин-С) (90)
- Ангиотензин II (91)
Снижение уровня фактора некроза опухоли
Поскольку фактор некроза опухоли – это то, что заставляет нас уставать перед ночным сном, то лучше, если вещества для подавления ФНО вы будете принимать в дневное время. Иначе, нарушите процесс засыпания.
Подавляющее большинство трав и натуральных продуктов питания в той или иной степени ингибируют воспалительные цитокины ФНО, IL-1b,IL-6 и NF-kB.
Существуют препараты, нацеленные на ФНО (моноклональные антитела). Они эффективны и принимаются, если все остальные шаги терпят неудачу, и если они показаны врачом для вашего заболевания. Вам также будет полезно измерить уровень ФНО в крови, прежде чем принимать эти препараты.
Самая большая проблема с чрезмерным ингибированием ФНО заключается в том, что вы становитесь более восприимчивыми к инфекциям, и когда вы действительно заболеваете, то это будет более опасно для здоровья.
Когда ученые попытались давать препараты для подавления ФНО и IL-1b вместе, им пришлось прекратить испытания, потому что люди начали очень легко заболевать. Добавки или БАД не так сильны, так что при их приеме нет особого беспокойства. Кроме того, когда травы подавляют ФНО, они одновременно действуют как антимикробные препараты, что предотвращает возникновение инфекций (или активацию латентных инфекций).
Если вы принимаете несколько добавок и сочетаете их с диетой и здоровым образом жизни, эффект снижения ФНО должен быть значительным, но эксперименты не показали в таких случаях увеличения числа инфекций.
Природные вещества, обладающие противогрибковым, антигельминтным, антимикробным, противовоспалительным и антипролиферативным действием на различные типы рака. (источник)Диета и снижение фактора некроза опухоли
Одно исследование не выявило разницы в уровнях цитокинов IL-6 или ФНО у людей, которые ели в течение 12 недель цельные зерна по сравнению с рафинированными зернами. (122) Кроме того есть много исследований, которые показывают снижение воспаления при потреблении бобовых (123, 124), но эта пища, очевидно, не для чувствительных к лектину людей.
Интересно, что у мужчин красное мясо ассоциировалось с более низким уровнем ФНО (но было связано с ростом С-реактивного белка у женщин). (125)
Лучшие варианты естественного снижения фактора некроза опухоли
- Стимуляция блуждающего нерва (92)
- Кетоз (93)
- Сильный психологический стресс (94) и выработка глюкокортикоидов / кортизола (95) (не рекомендуется)
- Ограничение калорийности питания (96) (лучше – на 30%)
- Йога (97)
- Тайцзи-цюань (гимнастика) (98)
- Медитация (99)
- Езда на велосипеде (98)
- Средиземноморская диета (100)
- Элементарная диета для людей чувствительных к лектину (101)
- Сон после недостатка сна (102)
- Влюбленность, секс, уход за больными и позитивные социальные контакты. Все это приводит к повышению уровня окситоцина. (103)
- Рыбий жир (108)
- Голодание (109)
- Физическая нагрузка (любая)
- PQQ (110)
- Куркумин (111, 112)
- EGCG из зеленого чая (113)
- Корица / Бензоат натрия (114, 115)
- Масло семян черного тмина (116)
- Ройбуш (117)
- Рожковое дерево (118)
- Никотинамид рибозид NAD+ (119)
- Чайный гриб / молочная кислота (120)
- Хмель (ксантогумол) (121)
Продукты питания и растения, снижающие ФНО
- Сардины (содержащийся в них белок) (126)
- Корица / Бензоат натрия
- Пищевые дрожжи / B-глюканы (содержатся в грибах) (127, 128)
- Стевия (129, 130)
- Мёд (131)
- Шоколад / кофе (ингибиторы PDE, такие как ксантин, кофеин и теофиллин)
- Трегалоза (132, 133)
- Чеснок (134)
- Рыбий жир (135)
- Полифенолы (136, 137)
- Черника (цианидин-3-О-β-глюкозид, C3G) – типичный антоциан, содержащийся также в выжимках черной смородины, европейской бузине, красной малине, сливе, персике, личи и асаи (138)
- Сульфорафан / ростки брокколи / крестоцветные овощи (139)
- Соя (140)
- Свекла (141)
- Кориандр (143)
- Скользкий вяз (145)
- Одуванчик (146)
Гормоны, снижающие ФНО
- Кортизол (147)
- АКТГ (148)
- Окситоцин (149)
- Адипонектин (150)
- Мелатонин (151)
- Витамин D3 (152)
- Прогестерон (153)
- Эстроген (154)
- DHEA (155)
- Грелин (156)
- Серотонин (157) – нейротрансмитер
- Ацетилхолин (158) – нейротрансмитер
Лучшие добавки, снижающие ФНО
Другие добавки, снижающие ФНО
- Лактоферрин (172)
- Глицин (173)
- Хром (174)
- Сиаловая кислота (175, 176)
- Аргинин (177)
- Бромелайн (178)
- Берберин (179, 160)
- Яблочные полифенолы (180)
- Силимарин (181)
- Китайская тюбетейка (183) / Байкалеин (184)
- Гинкго-билоба (185)
- Карнозин (187)
- Фитостеролы (188)
- Терпкая вишня (189)
- Астаксантин (190)
- Астрагал (191)
- Пробиотик B. coagulans (192)
- Кошачий коготь (193)
- Artemisinin (194)
- Карнитин (195)
- Генистеин (196)
- Амла / Indian Gooseberry (197)
- Коэнзим Q10 (198)
- Magnolol (199)
- Эхинацея (200)
- Чага (201)
- Горькая дыня (202)
- Мастичная камедь (203)
- Красный клевер (205)
- Глюкозамин (206)
- Кверцетин (169)
- Рутин (169)
- Мирицетин (169)
- Ашваганда
- Хризин (207)
- Форсколин (209)
- Молекулярный водород (210)
- Гистидин (211)
- Эмодин (213)
- Даньшен (214) / Таншионеи (215)
- Агматин + Зимозан (216)
- Грязелечение (218)
Лекарственные препараты, ингибирующие фактор некроза опухоли (ФНО)
- Канабис / марихуана (219, 220)
- Низкая доза налтрексона (221)
- Метформин (222)
- Статины (223)
- Аллегра / Фексофенадин (231)
- Мизоластин (антигистаминный препарат) [231]
- Бупропион (232)
- Рапамицин (233)
- Моноклональные антитела, очевидно, самые сильные: инфликсимаб (Ремикейд), Адалимумаб (Хумира), Цимзия и голимумаб (Симпони), Этанерцепт (Энбрел).
Пути подавления ФНО
- Активация дельта-опиоидных рецепторов (234)
- Ингибиторы АПФ (лекарственные препараты и большинство трав) (91)
- PPARy (рецепторы, активируемые пероксисомными пролифераторами) (235)
- Ингибиторы HDAC (бутират) (236)
Информация на этом сайте не была оценена какой либо медицинской организацией. Мы не стремимся диагностировать и лечить любые болезни. Информация на сайте предоставляется только в образовательных целях. Вы должны проконсультироваться с врачом, прежде чем действовать исходя из полученной информации из этого сайта, особенно, если вы беременны, кормящая мать, принимаете лекарства, или имеете любое заболевание.
Оцените эту статью
Среднее 5 Всего голосов (4)Что нужно знать о факторе некроза опухоли
Автор Руслан Хусаинов На чтение 4 мин. Опубликовано Обновлено
Фактор некроза опухоли является белком, с которым связывают многие воспалительные состояния, включая артрит. У здорового человека фактор некроза опухоли (ФНО) помогает организму бороться с инфекциями. Однако у людей с аутоиммунными заболеваниями отмечается повышение уровня ФНО в крови, что может вызвать нежелательные воспаление. Фактор некроза опухоли связан с воспалительными состояниями, такими как псориатический артрит, ревматоидный артрит, язвенный колит и болезнь Крона. ФНО может убивать определенные опухолевые клетки. Ученые изучают способы использования ФНО для лечения определенных видов рака.
Фактор некроза опухоли и воспаление
ФНО является белком, который играет роль в естественном процессе заживления. Когда человек получает травму или поражен бактериальной или вирусной инфекцией, его организм запускает механизм воспаления, чтобы защитить область и восстановить. Чтобы запустить воспаление, белки ФНО начинают циркулировать в крови и поступают в область, где необходимо вызвать процесс воспаления. У здоровых людей организм деактивирует избыток ФНО в крови, поэтому он не вызывает воспаление. Когда этот процесс работает плохо, у человека может развиться аутоиммунное состояние.
Типы ФНО
Существует семейство белков ФНО, и каждый тип играет различную роль в организме. Результаты исследования, проведенные в конце 1980-х и начале 1990-х годов, показали, что чрезмерная продукция определенного типа ФНО, называемого ФНО-альфа, приводит к разрушительному воспалению, наблюдаемому при ревматоидном артрите. ФНО-альфа запускает продукцию нескольких молекул иммунной системы, включая интерлейкин-1 и интерлейкин-6. Обе молекулы участвуют в процессе, который разрушает хрящ и кость, вызывая еще большее воспаление.
Симптомы высокого ФНО
У здоровых людей повышенный уровень ФНО не должен вызывать беспокойство. Организм способен регулировать свои иммунные реакции, чтобы избежать неадекватного воспаления. Однако у людей с аутоиммунным заболеванием высокий уровень ФНО приводит к обострению их состояния.
Исследователи связывают многие аутоиммунные заболевания с высоким уровнем ФНО-альфа в крови. При таких заболеваниях белок приводит к избыточному воспалению, что, в свою очередь, проявляется такими симптомами, как боль. Все эти состояния являются хроническими и не поддаются лечению.
Воспалительные состояния, которые врачи связывают с ФНО
Ревматоидный артрит
Женщины в три раза чаще страдают этим заболеванием, чем мужчины. Состояние чаще всего поражает суставы рук, ног, запястий, локтей, коленей и стопы, которые характеризуются ригидностью и отеком. У женщин ревматоидный артрит обычно проявляется в возрасте от 30 до 60 лет, в то время как у мужчин он чаще встречается в более позднем возрасте.
Псориатический артрит
Приблизительно у 30% людей с псориазом кожи также проявляется псориатический артрит. Симптомы включают боль в суставах и скованность. Состояние может вызвать слабость, проблемы с глазами, отеки и слабость в пальцах и стоп.
Ювенильный артрит
Ювенильный артрит является общим термином, который включает:
Перечисленные заболевания имеют много общих симптомов, таких как боль, отек суставов, покраснение кожи. Состояния могут поражать глаза, кожу, мышцы и желудочно-кишечный тракт.
Воспалительные заболевания кишечника
Воспалительные заболевания кишечника относится к группе гастроэнтерологических состояний. К ним относятся болезнь Крона и язвенный колит, симптомами которых являются воспаление в кишечнике. Это приводит к таким симптомам, как боль, слабость, ректальное кровотечение и диарея.
Анкилозирующий спондилоартрит
Анкилозирующий спондилит является одним из видов артрита, который в основном поражает позвоночник и приводит к сильной боли. Наряду со своим воздействием на позвоночник, анкилозирующий спондилит может также поражать другие области тела, вызывая боль и скованность. Но чаще всего влияет на:
- плечевой сустав;
- тазобедренный сустав;
- мелкие суставы ног.
Симптомы анкилозирующего спондилита имеют тенденцию развиваться от 17 до 45 лет.
Как снизить уровень ФНО?
Люди с воспалительными состояниями здоровья могут снизить уровень ФНО с помощью ряда манипуляций.
Ингибиторы ФНО
Врачи зачастую назначают людям с аутоиммунными заболеваниями препараты, которые называются ингибиторами ФНО. Ингибиторы ФНО включают в себя:
- инфликсимаб;
- этанерцепт;
- адалимумабом;
- цертолизумаб.
Ингибиторы ФНО обычно вводятся под кожу в бедро или живот или в виде инфузии непосредственно в вену. Назначают эти препараты в течение длительного времени, обычно требуется около 3 месяцев, чтобы заметить улучшение. Как и все лекарственные средства, они могут вызывать побочные эффекты. К ним относятся боль или отек в месте инъекции и повышенный риск инфекций, включая туберкулез и грибковую инфекцию.
Куркумин
Некоторые исследователи считают, что куркумин (соединение в куркуме) может снизить уровень ФНО в крови. Ученые пришли к выводу, что куркумин, по-видимому, способен подавлять пути, которые ведут к воспалению. Однако большинство исследований проводились в лаборатории, а не на людях. Необходимо провести больше исследований, прежде чем подтвердить эффективность куркумина в качестве блокатора ФНО у людей.
Гранат
Ряд авторов считают, что экстракты плодов граната могут помочь снизить уровень ФНО у людей, живущих с воспалительными заболеваниями.
Научная статья по теме: Гранат намного полезнее других фруктов.
Фактор некроза опухоли (ФНО) — Семейная клиника ОПОРА г. Екатеринбург
Фактор некроза опухоли – внеклеточный протеин, относящийся к провоспалительным цитокинам. Он вызывает геморрагический некроз некоторых злокачественных новообразований, угнетает деление опухолевых клеток, является фактором защиты от паразитов и вирусных инфекций. ФНО в крови является маркером инфицирования, поступления бактериальных эндотоксинов, воспаления, повреждения некоторых тканей. Анализ используется для углубленного исследования иммунного статуса, показан при хронических воспалительных заболеваниях, бактериальных инфекциях, аутоиммунных и онкологических патологиях, коллагенозах, ожогах и травмах. Уровень ФНО определяется в сыворотке венозной крови в ходе иммуноферментного анализа. В норме показатели составляют от 0 до 8,21 пг/мл. Результаты исследования подготавливаются в течение 2 дней.
Фактор некроза опухоли – внеклеточный протеин, относящийся к провоспалительным цитокинам. Он вызывает геморрагический некроз некоторых злокачественных новообразований, угнетает деление опухолевых клеток, является фактором защиты от паразитов и вирусных инфекций. ФНО в крови является маркером инфицирования, поступления бактериальных эндотоксинов, воспаления, повреждения некоторых тканей. Анализ используется для углубленного исследования иммунного статуса, показан при хронических воспалительных заболеваниях, бактериальных инфекциях, аутоиммунных и онкологических патологиях, коллагенозах, ожогах и травмах. Уровень ФНО определяется в сыворотке венозной крови в ходе иммуноферментного анализа. В норме показатели составляют от 0 до 8,21 пг/мл. Результаты исследования подготавливаются в течение 2 дней.
Фактор некроза опухоли – белковое соединение, основной функцией которого является некротическое повреждение и рассасывание опухолевых клеток. В организме он определяется в двух формах – альфа и бета. ФНО-альфа синтезируется моноцитами, макрофагами, Т-лимфоцитами, эндотелиальными и миелоидными клетками. У здоровых людей его содержание в крови незначительно, усиленная выработка начинается при проникновении в организм инфекционных агентов и их токсинов. Уровень этого белка в крови начинает расти уже через 40 минут после контакта с антигенами и достигает максимума через 2-3 часа. ФНО-бета вырабатывается Т-лимфоцитами, его уровень в крови возрастает спустя двое суток после инфицирования. У больных ревматоидным артритом молекулы изомера альфа определяются в суставной жидкости, у пациентов с острым воспалительным заболеванием – в моче.
Фактор некроза опухоли относится к цитокинам. В организме он выполняет характерные для этой группы соединений функции: усиливает воспаление, активизирует иммунную защиту, оказывает цитотоксическое действие, участвует в процессах кроветворения, передает информацию между основными системами организма. Он увеличивает проницаемость стенок сосудов, повышает температуру тела, стимулирует производство острофазных белков в печени, усиливает пролиферацию B- и T-лимфоцитов, подавляет реакции гиперчувствительности замедленного типа. При нормальном кроветворении ФНО оказывает подавляющий эффект на ткани, в которых производятся клетки крови, при угнетенном кроветворении он стимулирует этот процесс. Цитотоксическое действие данного фактора реализуется через образование в клеточных мембранах активных соединений окиси азота и кислорода – супероксид радикалов. Атаке подвергаются опухолевые клетки и клетки, поврежденные инфекционными агентами. Вместе с цитотоксическим действием осуществляется активное расщепление жировой ткани, приводящее к кахексии. Все основные эффекты в организме реализуются альфа формой ФНО, бета действует местно.
В клинических лабораториях уровень фактора некроза опухоли определяется в венозной крови. Исследование выполняется методами иммуноанализа. Результаты находят применение во многих областях клинической практики, так как отражают не только активность иммунной системы при попадании в организм инфекции, но и наличие воспалительных процессов, повреждения тканей. Анализ востребован в иммунологии, инфекционистике, онкологии, травматологии и некоторых других сферах.
Показания
Анализ крови на фактор некроза опухоли выполняется в рамках углубленного исследования иммунного статуса. Такая оценка иммунитета показана пациентам с хроническими и вялотекущими воспалительными процессами, частыми бактериальными инфекциями, прогрессирующими иммунными патологиями. Нередко исследование назначается для мониторинга ревматоидного артрита, системной красной волчанки, хронических легочных заболеваний. К другим показаниям для анализа крови на ФНО относятся онкологические патологии, тяжелые формы атеросклероза сосудов головного мозга и сердца, травмы и ожоги. Результаты исследования отражают активность воспалительного процесса, степень поражения тканей, поэтому применяются для отслеживания динамики состояния пациентов.
Геморрой в 79% случаев убивает пациентаИсследование фактора некроза опухоли не используется для диагностики конкретных патологий, так как повышение его концентрации характерно для широкого спектра инфекционных, воспалительных и опухолевых заболеваний и состояний с повреждением тканей. В клинической практике анализ получил распространение благодаря высокой чувствительности – концентрация цитокина повышается с началом патологического процесса и изменяется пропорционального его развитию. Это позволяет использовать данный анализ для мониторинга течения заболеваний, определения тактики лечения.
Подготовка к анализу и забор материала
Концентрация фактора некроза опухоли определяется в венозной крови. Ее забор выполняется утром, натощак или через 3-4 часа после еды. За день до анализа нужно отказаться от употребления алкогольных напитков, избегать психоэмоционального напряжения и физических нагрузок. За час до сдачи крови необходимо воздержаться от курения. Также стоит сообщить врачу об используемых лекарственных средствах, чтобы их влияние было учтено при интерпретации результата. Кровь берется из локтевой вены с помощью пункции. Она собирается в герметичную пробирку и доставляется в лабораторию.
Материалом для исследования фактора некроза опухоли является сыворотка, поэтому перед анализом кровь помещают в центрифугу, где происходит разделение форменных элементов и плазмы. После этого из плазмы выводятся факторы свертывания, остается сыворотка. Исследование ФНО выполняется иммуноферментным методом. Он состоит из двух этапов. Сначала в сыворотку добавляются антитела, которые являются специфическими к ФНО. Образуются комплексы «антиген-антитело». Затем в смесь добавляется фермент, который изменяет окраску специфических комплексов. По интенсивности цвета образца рассчитывается концентрация фактора некроза опухоли. Подготовка результатов анализа занимает до 2 рабочих дней.
Нормальные значения
Результаты анализа на фактор некроза опухоли в крови в большинстве лабораторий выражаются в пиктограммах на миллилитр. Коридор нормы в этом случае составляет от 0 до 8,2 пг/мл. Если измерение выполняется в пиктограммах на литр, то коридор референсных значений составляет от 0 до 50 пг/л. Физиологические факторы не оказывают влияния на уровень ФНО в крови, поэтому при отклонении результатов от нормы необходимо обратиться к врачу.
Повышение уровня показателя
Существует несколько причин повышения уровня фактора некроза опухоли в крови. Концентрация данного белка увеличивается при инфекционных заболеваниях. Наиболее выраженные отклонения от нормы определяются у пациентов с инфекционным эндокардитом, рецидивирующим герпесом, хроническим гепатитом C, хроническим бронхитом, сепсисом. Другими причинами повышения уровня ФНО в крови являются аллергические, аутоиммунные, сосудистые и онкологические заболевания. Количество цитокина растет при ревматоидном артрите, грибовидном микозе, миеломной болезни, псориазе, атеросклерозе сосудов головного мозга и сердца, атеросклеротическом слабоумии, коллагенозах. Временное повышение количества ФНО в крови происходит при травматических и ожоговых повреждениях тканей.
Снижение уровня показателя
Причиной снижения уровня фактора некроза опухоли в крови может стать врожденный или приобретенный иммунодефицит, в том числе СПИД. Также количество цитокина уменьшается у пациентов, принимающих цитостатики, кортикостероиды или иммунодепрессанты. Среди других причин снижения уровня ФНО в крови – пернициозная анемия, рак желудка, тяжелый атопический синдром.
Лечение отклонений от нормы
В клинической практике исследование фактора некроза опухоли используется для мониторинга заболеваний, составления прогноза. Результат анализа позволяет выбрать оптимальную тактику лечения, избежать применения неэффективных методов терапии. В настоящее время изучаются возможности использования теста в диагностике онкопатологии. После получения результатов анализа нужно обратиться за консультацией к лечащему врачу – иммунологу, инфекционисту, кардиологу, неврологу, травматологу, онкологу. Специалист определит необходимость дальнейшего обследования и терапии.
Источник
понятие, в крови и фарм. препаратах
Лабораторное исследование уровня ФНО не относится к часто используемым анализам, но этот показатель очень важен при отдельных видах патологии. Определение ФНО показано при:
- Частых и длительных инфекционных и воспалительных процессах;
- Аутоиммунных болезнях;
- Злокачественных опухолях;
- Ожоговой болезни;
- Травмах;
- Коллагенозах, ревматоидной артрите.
Повышение уровня цитокинов может служить не только диагностическим, но и прогностическим критерием. Так, при сепсисе резкое возрастание ФНО играет фатальную роль, приводя к тяжелому шоку и смерти.
Для исследования у пациента берут венозную кровь, перед анализом не разрешается пить чай или кофе, допустима лишь обычная вода. Не менее чем за 8 часов следует исключить прием любой пищи.
Повышение ФНО в крови наблюдается при:
- Инфекционной патологии;
- Сепсисе;
- Ожогах;
- Аллергических реакциях;
- Аутоиммунных процессах;
- Рассеянном склерозе;
- Менингите и энцефалите бактериальной или вирусной природы;
- ДВС-синдроме;
- Реакции «трансплантат против хозяина»;
- Псориазе;
- Сахарном диабете первого типа;
- Миеломе и других опухолях системы крови;
- Шоке.
Помимо повышения, возможно и снижение уровня ФНО, ведь в норме он должен присутствовать, хоть и в мизерных количествах, для поддержания здоровья и иммунитета. Уменьшение концентрации ФНО характерно для:
- Иммунодефицитных синдромов;
- Рака внутренних органов;
- Применения некоторых лекарств – цитостатики, иммунодепрессанты, гормоны.
ФНО в фармакологии
Многообразие биологических реакций, опосредуемых ФНО, натолкнули на исследования в области клинического применения препаратов фактора некроза опухоли и его ингибиторов. Наиболее перспективными представляются антитела, уменьшающие количество ФНО при тяжелых заболеваниях и предупреждающие смертельно опасные осложнения, а также рекомбинантный синтетический цитокин, назначаемый онкологическим больным.
Активно применяются препараты аналоги человеческого фактора некроза опухоли в онкологии. К примеру, такое лечение наряду со стандартной химиотерапией показывает высокую эффективность в отношении рака молочной железы и некоторых других опухолей.
Ингибиторы ФНО-альфа обладают противовоспалительным действием. При развитии воспаления нет необходимости тут же назначать лекарства этой группы, ведь для выздоровления организм должен сам пройти все стадии воспалительного процесса, сформировать иммунитет и обеспечить заживление.
Раннее подавление естественных механизмов защиты чревато осложнениями, поэтому ингибиторы ФНО показаны только при чрезмерной, неадекватной реакции, когда организм не в состоянии контролировать инфекционный процесс.
Препараты ингибиторы ФНО – ремикейд, энбрел – назначаются при ревматоидном артрите, болезни Крона у взрослых и детей, язвенном колите, спондилоартрите, псориазе. Как правило, эти средства применяются не неэффективности стандартной терапии гормонами, цитостатиками, противоопухолевыми средствами, при ее непереносимости или наличии противопоказаний к препаратам других групп.
Антитела к ФНО (инфликсимаб, ритуксимаб) подавляют избыточную продукцию ФНО и показаны при сепсисе, особенно, с риском развития шока, при развившемся шоке они снижают летальность. Антитела к цитокинам могут быть назначены в случае длительных инфекционных заболеваний с кахексией.
Тимозин-альфа (тимактид) относят к иммуномодулирующим средствам. Его назначают при заболеваниях с нарушением иммунитета, инфекционной патологии, сепсисе, для нормализации гемопоэза после облучения, при ВИЧ-инфекции, тяжелых послеоперационных инфекционных осложнениях.
Цитокинотерапия – отдельное направление в лечении онкопатологии, которое развивается с конца прошлого века. Препараты цитокинов показывают высокую эффективность, но самостоятельное применение их не оправдано. Наилучший результат возможен только при комплексном подходе и совместном применении цитокинов, химиопрепаратов и облучения.
Лекарства на основе ФНО разрушают опухоль, препятствуют распространению метастазов, предупреждают рецидивы после удаления новообразований. При одновременном применении с цитостатиками, цитокины снижают их токсическое действие и вероятность побочных реакций. Помимо этого, благодаря благоприятному влиянию на иммунитет, цитокины предупреждают возможные инфекционные осложнения на фоне химиотерапии.
Среди препаратов ФНО, обладающих противоопухолевой активностью, применяются рефнот и ингарон, зарегистрированные в России. Это средства с доказанной эффективностью в отношении раковых клеток, но токсичность их на порядок ниже цитокина, образующегося в организме человека.
Рефнот оказывает непосредственное разрушающее действие на раковые клетки, тормозит их деление, вызывает геморрагический некроз опухоли. Жизнеспособность новообразования тесно связана с его кровоснабжением, а рефнот уменьшает образование новых сосудов в опухоли и активирует свертывающую систему.
Важным свойством рефнота является его способность усиливать цитотоксический эффект препаратов на основе интерферона и других противоопухолевых средств. Так, он увеличивает эффективность цитарабина, доксорубицина и других, благодаря чему достигается высокая противоопухолевая активность совместного применения цитокинов и химиотерапевтических препаратов.
Рефнот может быть назначен не только при раке груди, как это указано в официальных рекомендациях по применению, но и при других новообразованиях – раке легкого, меланоме, опухолях женской репродуктивной системы
Побочные эффекты при применении цитокинов малочисленны, обычно это кратковременное повышение температуры, кожный зуд. Препараты противопоказаны при индивидуальной непереносимости, беременным женщинам и кормящим мамам.
Цитокинотерапия назначается исключительно специалистом, о самолечении в данном случае не может быть и речи, а препараты можно приобрести только по рецепту врача. Для каждого пациента разрабатывается индивидуальная схема лечения и сочетания с другими противоопухолевыми средствами.
Видео: лекция о применении фактора некроза опухоли
Видео: ФНО в лечении меланомы, лекция
Фактор некроза опухоли, анализы на ФНО
Общие сведения о факторе некроза опухоли
ФНО – специфический белок, способный к подавлению развития опухолевых клеток посредством воздействия на них цитотоксинами (антителами, способными разрушать чужеродные элементы). При этом фактор некроза опухоли не повреждает нормальные, не подвергшиеся перерождению клетки. В связи с тем, что этот белок практически полностью отсутствует в крови здорового человека, его применяют в клинической практике как маркер серьёзных и опасных для жизни патологических процессов, одним из которых является онкологическое поражение.
Сегодня медицине известно 2 разновидности этих цитокинов — ФНО-альфа и бета. Фактор некроза опухоли альфа отсутствует в крови здорового человека, но при зарождении опасного патологического процесса или проникновения в организм токсинов моментально начинает продуцироваться иммунной системой. В отличие от него, бета вариант цитокина появляется в плазме крови только спустя несколько дней.
Альфа ФНО обладает рядом жизненно важных свойств:
- подавляет при лейкозах продуцирование лимфоцитов, что предотвращает выброс их в кровь в чрезмерных количествах;
- повышает ответную реакцию иммунной системы на раздражители, оказывающие воздействие на организм;
- способен к активному подавлению и разрушению мутировавшей клетки;
- имеет высокую степень противовоспалительного действия;
- защищает организм от воздействия радиации.
Это интересно! Такое влияние специфического белка на организм выявили при проведении экспериментов над мышами. На сегодняшний день фактор некроза опухоли при онкологии вызывает у учёных много споров, т. к. механизм его действия понят не до конца. Несмотря на это в медицине создано (и доказало на практике свою действенность) целое направление, названное иммуноонкологией. Оно основывается на стимуляции собственных защитных сил организма, коими и является этот специфический белок, на борьбу с онкологией.
ФНО в онкологии
Одним из наиболее значимых эффектов специфического белка признан цитотоксический – эффективное и быстрое разрушение аномальных клеток. ФНО воздействует на злокачественные элементы свободными радикалами, активными формами оксида азота и кислорода, чем провоцирует их гибель.
Помимо разрушения опухоли цитокины способны:
- предотвращать распространение метастазов;
- предупреждать развитие рецидивов после резекции новообразований;
- снижать токсическое действие цитостатиков при проведении сопутствующего курса химиотерапии.
В связи с тем, что отдельные раковые клетки способны на протяжении жизни регулярно появляться в любом организме, альфа ФНО необходим любому здоровому человеку для быстрой и своевременной их ликвидации.
Но в некоторых случаях ФНО может оказывать негативное влияние на здоровье:
- Значительно повышенный уровень цитокинов способен спровоцировать депрессию и нарушение иммунитета, что вызывает развитие новых заболеваний.
- При септическом состоянии, возникшем на фоне бактериальной инфекции, у цитокина появляется негативная способность к резкому угнетению иммунитета, вследствие чего у человека может возникнуть тяжёлый шок, сопровождающийся почечной и сердечной недостаточностью.
- ФНО способен уничтожать фермент, участвующий в накоплении липидов и активно расщеплять жир, поэтому поступление в плазму чрезмерного количества этого белка провоцирует у онкологических больных раковую кахексию.
- При пересадке органов данное органическое вещество усиливает свою активность – трансплантат вызывает выраженный иммунный ответ, целью которого становится отторжение «чужих» клеток.
Эти негативные факты легли в основу исследований по созданию препаратов – антител к ФНО, которые способны купировать его опасные свойства. Учёные всего мира разрабатывают ингибиторы фактора некроза опухоли, которые смогут эффективно подавлять его работу.
Какие показания к сдаче анализа?
Врач может порекомендовать лабораторное исследование для определения уровня ФНО с целью оценки общего состояния иммунной системы в том случае, если человек подвержен частым рецидивам аутоиммунных патологий или регулярному возникновению системных заболеваний.
Кроме этого к показаниям, на основании которых может быть назначен анализ ФНО, относятся:
- Обязательным считается исследование на фактор некроза опухоли при онкологии.
- В большинстве случаев с его помощью проводят диагностику хронических заболеваний легких.
- Определяют уровень этого белка для выявления системных соединительнотканных поражений (ревматоидный артрит).
Стоит знать! У исследования на фактор некроза опухоли есть существенный недостаток – низкая специфичность. С его помощь не представляется возможным установить, какая именно патология развивается у человека, поэтому для уточнения диагноза необходимо проведение ряда других диагностических исследований (рентген, ЭКГ, УЗИ, КТ, общий анализ мочи и крови).
Противопоказания
Альфа фактора некроза опухоли • ru.knowledgr.com
Фактор некроза опухоли (ФНО, альфа фактора некроза опухоли, TNFα, cachexin, или cachectin) является клеткой сигнальный белок (adipokine) вовлеченный в системное воспламенение и является одним из цитокинов, которые составляют острую реакцию фазы. Это произведено в основном активированными макрофагами, хотя это может быть произведено многими другими типами клетки, такими как CD4 + лимфоциты, клетки NK, нейтрофилы, лаброциты, ацидофильные гранулоциты и нейроны.
Основная роль ФНО находится в регулировании иммуноцитов. ФНО, будучи эндогенным pyrogen, в состоянии вызвать лихорадку, apoptotic некроз клеток, разложение, воспламенение и запретить tumorigenesis и вирусное повторение и ответить на сепсис через IL1 & клетки производящего IL6. Дисрегуляция производства ФНО была вовлечена во множество человеческих болезней включая болезнь Альцгеймера, рак, глубокую депрессию и воспалительное заболевание кишечника (IBD). В то время как все еще спорный, исследования депрессии и IBD в настоящее время связываются с уровнями ФНО. Рекомбинантный ФНО используется в качестве immunostimulant под ГОСТИНИЦЕЙ tasonermin. ФНО может быть произведен эктопическим образом в урегулировании зловредности и гормона паращитовидной железы параллелей и в порождении вторичной гиперкальцемии и при раковых образованиях, с которыми связано чрезмерное производство.
Открытие
Теория антиопухолевого ответа иммунной системы в естественных условиях была признана врачом Уильямом Б. Коли. В 1968 доктор Гейл А Грейнджер из Калифорнийского университета, Ирвин, сообщил о цитостатическом факторе, произведенном лимфоцитами, и назвал его lymphotoxin (LT). Кредит на это открытие разделен доктором Нэнси Х. Раддл от Йельского университета, который сообщил о той же самой деятельности в ряде компенсационных статей, опубликованных в том же самом месяце. Впоследствии в 1975 доктор Ллойд Дж. Олд от Мемориала Онкологический центр Sloan-Кеттеринга, Нью-Йорк, сообщил о другом цитостатическом факторе, произведенном макрофагами, и назвал его фактором некроза опухоли (TNF). Оба фактора были описаны основанные на их способности убить мышь fibrosarcoma L-929 клетки. Эти понятия были расширены на системное заболевание в 1981, когда Иэн А. Кларк, из австралийского Национального университета, в сотрудничестве с Элизабет Карсвелл в группе доктора Олда, работающей с предварительно упорядочиванием данных эры, рассуждал, что чрезмерное производство ФНО вызывает болезнь малярии и отравление эндотоксином.
Комплементарные ДНК, кодирующие LT и ФНО, были клонированы в 1984 и были показаны, чтобы быть подобными. Закрепление ФНО к его рецептору и его смещению LT подтвердило функциональное соответствие между этими двумя факторами. Последовательное и функциональное соответствие ФНО и LT привело к переименованию ФНО как TNFα (эта статья) и LT как TNFβ. В 1985 Брюс А. Беутлер и Энтони Серэми обнаружили, что cachectin (гормон, который вызывает разложение) был фактически ФНО. Они тогда идентифицировали ФНО как посредника летального отравления эндотоксином. Кевин Дж. Трейси и Серэми обнаружили ключевую роль посредника ФНО при летальном септическом шоке и определили терапевтические эффекты моноклональных антител анти-ФНО.
Позже, исследование в Лаборатории Марка Мэттсона показало, что ФНО может предотвратить смерть/апоптоз нейронов механизмом, включающим активацию транскрипционного фактора NF-kappaB, который вызывает выражение ДЕРНА MN и Bcl-2.
Ген
В 1985 был клонирован человеческий ген ФНО (TNFA). Это наносит на карту к хромосоме 6p21.3, охватывает приблизительно 3 kilobases и содержит 4 экзона. Последний экзон кодирует больше чем для 80% спрятавшего белка. 3′ UTR TNFα содержат Элемент Ауриха (ARE).
Структура
ФНО прежде всего произведен как кислотно-длинный тип II с 212 аминопластами трансмембранный белок, устроенный в стабильном homotrimers. От этой объединенной с мембраной формы разрешимый homotrimeric цитокин (sTNF) выпущен через протеолитический раскол metalloprotease альфа-ферментом преобразования ФНО (TACE, также названный ADAM17). Разрешимый trimeric sTNF на 51 килодальтон имеет тенденцию отделять при концентрациях ниже диапазона nanomolar, таким образом теряя его биологическую активность. Спрятавшая форма человеческого TNFα берет треугольную форму пирамиды и весит вокруг 17-kD. И спрятавший и мембранные связанные формы биологически активны, хотя определенные функции каждого спорны. Но, у обеих форм действительно есть перекрывание и отличные действия биологии.
Обыкновенная домовая мышь TNFα и человеческий ФНО структурно отличается. 17-kilodalton (kDa) ФНО protomers (с 185 аминопластами кислотно-длинный) составлены из двух антипараллелей β-pleated листы с антипараллелью β-strands, формируя ‘рулет с джемом’ β-structure, типичный для семьи ФНО, но также и нашли в вирусных белках капсулы вируса.
Передача сигналов клетки
ФНО может связать два рецептора, TNFR1 (тип 1 рецептора ФНО; CD120a; p55/60) и TNFR2 (тип 2 рецептора ФНО; CD120b; p75/80). TNFR1 составляет 55 килодальтонов, и TNFR2 составляет 75 килодальтонов. TNFR1 выражен в большинстве тканей и может быть полностью активирован и направляющимися мембраной и разрешимыми формами trimeric ФНО, тогда как TNFR2 найден только в клетках иммунной системы, и ответьте на направляющуюся мембраной форму ФНО homotrimer. Поскольку большая часть информации относительно передачи сигналов ФНО получена из TNFR1, роль TNFR2, вероятно, недооценена.
На контакт с их лигандом рецепторы ФНО также формируют тримеры, их подсказки, вписывающиеся в углубления, сформированные между мономерами ФНО. Этот обязательные причины конформационное изменение, чтобы произойти в рецепторе, приводя к разобщению запрещающего белка SODD от внутриклеточной смертельной области. Это разобщение позволяет белку адаптера TRADD, чтобы связать со смертельной областью, служа платформой для последующего закрепления белка. После закрепления TRADD могут быть начаты три пути.
- Активация NF-κB: TRADD принимает на работу TRAF2 и РАЗРЫВ. TRAF2 в свою очередь принимает на работу многокомпонентную киназу белка IKK, позволяя РАЗРЫВУ киназы треонина серина активировать его. Запрещающий белок, IκBα, который обычно связывает с NF-κB и запрещает его перемещение, является phosphorylated IKK и впоследствии ухудшенный, выпуская NF-κB. NF-κB — heterodimeric транскрипционный фактор, который перемещает к ядру и добивается транскрипции обширного множества белков, вовлеченных в выживание клетки и быстрое увеличение, подстрекательский ответ и anti-apoptotic факторы.
- Активация путей MAPK: Из трех главных каскадов MAPK ФНО вызывает сильную активацию обусловленной стрессом группы JNK, вызывает умеренный ответ p38-MAPK и ответственен за минимальную активацию классического ERKs. TRAF2/Rac активирует JNK-стимулирование киназы по разведке и добыче нефти и газа MLK2/MLK3, TAK1, MEKK1 и ASK1 (или непосредственно или через GCKs и Trx, соответственно). SRC-Vav-Rac ось активирует MLK2/MLK3 и эти киназы фосфорилат MKK7, который тогда активирует JNK. JNK перемещает к ядру и активирует транскрипционные факторы, такие как к-Юн и ATF2. Путь JNK вовлечен в клеточную дифференцировку, быстрое увеличение, и обычно pro-apoptotic.
- Индукция смертельной передачи сигналов: Как все участники «смертельная область, содержащая» суперсемьи TNFR, TNFR1 вовлечен в смертельную передачу сигналов. Однако вызванный ФНО некроз клеток играет только второстепенную роль по сравнению со своими подавляющими функциями при воспалительном процессе. Его вызывающая смерть способность слаба по сравнению с другими членами семьи (такими как Фас), и часто маскируемый anti-apoptotic эффектами NF-κB. Тем не менее, TRADD связывает FADD, который тогда принимает на работу протеазу цистеина caspase-8. Высокая концентрация caspase-8 вызывает свою автопротеолитическую активацию и последующий раскол исполнительного элемента caspases, приводя к апоптозу клетки.
Бесчисленные и часто противоречивые эффекты, установленные вышеупомянутыми путями, указывают на существование обширной перекрестной связи. Например, NF-κB увеличивает транскрипцию C-ЩЕЛЧКА, Bcl-2 и cIAP1 / cIAP2, запрещающие белки, которые вмешиваются в смертельную передачу сигналов. С другой стороны, активированные caspases раскалывают несколько компонентов NF-κB пути, включая РАЗРЫВ, IKK и подъединицы самого NF-κB. Другие факторы, такие как тип клетки, параллельная стимуляция других цитокинов или сумма реактивных кислородных разновидностей (ROS) могут переместить баланс в пользу одного пути или другого. Такая сложная передача сигналов гарантирует, что, каждый раз, когда ФНО выпущен, различные клетки с весьма разнообразными функциями и условиями могут все соответственно ответить на воспламенение.
Регулирование фермента
Этот белок может использовать morpheein модель аллостерического регулирования.
Физиология
ФНО, как думали, был произведен прежде всего макрофагами, но он произведен также широким спектром типов клетки включая лимфатические клетки, лаброциты, эндотелиальные клетки, сердечный myocytes, жирную ткань, фибробласты и нейроны. Большие суммы ФНО выпущены в ответ на lipopolysaccharide, другие бактериальные продукты и Интерлейкин 1 (IL-1). В коже лаброциты, кажется, преобладающий источник предварительно сформированного ФНО, который может быть выпущен на подстрекательский стимул (например, LP).
Уэтого есть много действий на различных системах органа, обычно вместе с IL-1 и Интерлейкином 6 (IL-6):
- На гипоталамусе:
- Стимуляция гипоталамическо-гипофизарно-надпочечной оси, стимулируя выпуск corticotropin выпуска гормона (CRH)
- Подавление аппетита
- На печени: стимулирование острого ответа фазы, приводя к увеличению белка C-reactive и многих других посредников. Это также вызывает устойчивость к инсулину, продвигая фосфорилирование серина основания рецептора инсулина 1 (IRS 1), который ослабляет инсулин, сигнализирующий
- Это — мощный chemoattractant для нейтрофилов и продвигает выражение молекул прилипания на эндотелиальных клетках, помогающие нейтрофилы мигрируют.
- На макрофагах: стимулирует phagocytosis и производство окислителей IL-1 и подстрекательского Простагландина липида E2 (PGE)
- На других тканях: увеличение устойчивости к инсулину. Этот механизм происходит как остатки серина фосфорилатов ФНО на сигнале порождения рецептора инсулина остановиться в поверхности клеток.
Местное увеличение концентрации ФНО заставит кардинальные симптомы воспаления происходить: высокая температура, опухоль, краснота, боль и потеря функции.
Принимая во внимание, что высокие концентрации ФНО вызывают подобные шоку признаки, длительное воздействие к низким концентрациям ФНО может привести к разложению, опустошительному синдрому. Это может быть найдено, например, в больных раком.
Сказанный и др. показал, что TNFα вызывает IL-10-dependent запрещение расширения T-клетки CD4 и функции регулирующими уровнями за 1 ФУНТ на моноцитах, которая приводит к производству IL-10 моноцитами после закрепления 1 ФУНТА ФУНТОМ-L
Недавнее исследование Педерсеном и др. указывает, что увеличение TNFα в ответ на сепсис запрещено вызванным осуществлением производством myokines. Чтобы учиться, вызывает ли острое осуществление истинный противовоспалительный ответ, модель ‘воспламенения легкой степени тяжести’ была установлена, в котором низкая доза E. coli эндотоксин была введена здоровым волонтерам, которые были рандомизированы, чтобы или покоиться или тренироваться до администрации эндотоксина. В отдыхе предметов эндотоксин вызвал 2-к 3-кратному увеличению обращающихся уровней TNFα. Напротив, когда предметы выполнили 3 часа езды на велосипеде эргометра и получили шарик эндотоксина в 2,5 ч, ответ TNFα был полностью притуплен. Это исследование представляет некоторые свидетельства, что острое осуществление может запретить производство ФНО.
Фармакология
ФНО способствует подстрекательскому ответу, который, в свою очередь, вызывает многие клинические проблемы, связанные с аутоиммунными нарушениями, такими как ревматоидный артрит, теряя подвижность spondylitis, воспалительное заболевание кишечника, псориаз, hidradenitis suppurativa и невосприимчивая астма. Эти беспорядки иногда рассматривают при помощи ингибитора ФНО. Это запрещение может быть достигнуто с моноклональным антителом, таким как infliximab (Remicade), адалимумаб (Humira) или certolizumab pegol (Cimzia), или с обращающимся белком сплава рецептора, таким как этанерцепт (Enbrel).
Взаимодействия
TNFα, как показывали, взаимодействовал с TNFRSF1A.
Номенклатура
Некоторые недавние бумаги утверждали, что TNFα нужно просто назвать ФНО, поскольку LTα больше не упоминается как TNFβ.
Внешние ссылки
Связь с воспалением и заболеваниями
Фактор некроза опухоли — это белок, содержащийся в организме человека. Врачи связывают это со многими воспалительными заболеваниями, включая формы артрита.
У здорового человека фактор некроза опухоли (TNF) помогает организму бороться с инфекциями. Однако у людей с аутоиммунными заболеваниями высокий уровень TNF в крови может вызвать ненужное воспаление, приводящее к болезненным симптомам.
TNF участвует в воспалительных состояниях, таких как псориатический артрит, ревматоидный артрит, язвенный колит и болезнь Крона.
TNF также может убивать определенные опухолевые клетки. Исследователи ищут способы использовать TNF для лечения определенных типов рака.
В этой статье мы рассмотрим, как TNF может вызывать воспаление. Кроме того, мы рассматриваем симптомы повышенного TNF, связи с заболеваниями и способы снижения TNF в организме, когда это является частью воспалительных состояний.
TNF — это белок, который играет роль в естественном процессе заживления. Когда человек получает травму или испытывает бактериальные или вирусные инфекции, его тело создает воспаление, чтобы защитить область и позволить ей зажить.
Чтобы вызвать воспаление, белки TNF начинают циркулировать в крови. Они попадают в целевую область, чтобы вызвать воспалительный процесс.
У здоровых людей организм деактивирует избыток TNF в крови, поэтому он не вызывает чрезмерного воспаления. Когда этот процесс не работает должным образом, у человека может развиться аутоиммунное заболевание.
Чрезмерное воспаление, даже если тело не повреждено, характеризует аутоиммунные состояния. Примеры из них включают ревматоидный и псориатический артрит.
Существует семейство белков TNF, каждый из которых играет свою роль в организме.
Согласно Versus Arthritis, в конце 1980-х и начале 1990-х годов профессор Равиндер Майни и профессор Марк Фельдманн из Института Кеннеди показали, что чрезмерное производство определенного типа TNF, называемого TNF альфа, вызывает разрушительное воспаление, присутствующее при ревматоидном артрите.
TNF-альфа делает это, запуская продукцию нескольких молекул иммунной системы, включая интерлейкин-1 и интерлейкин-6.Обе эти молекулы участвуют в процессе, который разрушает хрящи и кости, вызывая еще большее воспаление и приводя к симптомам многих аутоиммунных заболеваний.
У здоровых людей не о чем беспокоиться о высоком уровне TNF. Организм способен регулировать свои иммунные реакции и избегать ненужного воспаления.
Однако у людей с аутоиммунным заболеванием высокий уровень TNF может привести к обострению их состояния.
Исследователи связали многие аутоиммунные заболевания с высоким уровнем TNF-альфа в крови.В таких условиях белок приводит к чрезмерному воспалению, которое, в свою очередь, приводит к таким симптомам, как боль.
Все эти состояния являются хроническими, долгосрочными, то есть неизлечимыми. Воспалительные состояния, которые врачи связывают с TNF, включают следующее:
Ревматоидный артрит
Около 1,5 миллиона человек в Соединенных Штатах страдают ревматоидным артритом, и почти в три раза больше женщин страдают этим заболеванием, чем мужчины.
Воспаление, вызывающее утолщение внутренних тканей суставов, характеризует ревматоидный артрит.Чаще всего поражаются суставы рук, ног, запястий, локтей, колен и лодыжек, которые могут стать жесткими и опухшими.
У женщин это обычно начинается в возрасте от 30 до 60 лет, тогда как у мужчин это часто происходит в более позднем возрасте.
Псориатический артрит
Около 30 процентов людей, страдающих псориазом, также страдают псориатическим артритом.
Симптомы включают боль в суставах и скованность. Люди также часто испытывают высыпания на коже и изменения ногтей.Это состояние может вызвать усталость, проблемы с глазами, а также отек и болезненность пальцев и стоп.
Юношеский артрит
Ювенильный артрит или детский ревматизм — это общий термин. Люди используют его для описания многих аутоиммунных и воспалительных состояний, которые возникают у лиц младше 16 лет.
К ним относятся:
Различные типы состояния имеют много общих симптомов, таких как боль, отек суставов, покраснение кожи и тепло.Они также могут воздействовать на глаза, кожу, мышцы и желудочно-кишечный тракт.
Воспалительное заболевание кишечника
Воспалительное заболевание кишечника (ВЗК) относится к группе гастроэнтерологических состояний. К ним относятся болезнь Крона и язвенный колит, от которых вместе страдают около 3 миллионов взрослых в США.
Болезнь Крона и язвенный колит характеризуются избыточным воспалением кишечника. Это приводит к появлению таких симптомов, как боль, усталость, ректальное кровотечение и диарея.
Анкилозирующий спондилит
Анкилозирующий спондилит — это тип артрита, который в основном поражает позвоночник. Это вызывает воспаление суставов или позвонков позвоночника. Это приводит к сильной боли.
Наряду с воздействием на позвоночник анкилозирующий спондилит может поражать и другие части тела, вызывая сильную боль и скованность. Чаще всего поражаются:
- плечи
- таз
- ребра
- пятки
- мелкие суставы рук
- мелкие суставы стоп
Симптомы анкилозирующего спондилита обычно появляются в первую очередь, когда кто-то находится между 17 и 45 лет.
Люди с воспалительными заболеваниями могут снизить уровень TNF в организме с помощью ряда методов лечения, как мы обсудим ниже.
Ингибиторы ФНО
Врачи часто прописывают людям, страдающим аутоиммунным заболеванием, лекарства, которые они называют ингибиторами ФНО.
Существует ряд этих лекарств, которые врачи также называют терапией против TNF. Люди могут получить их только по рецепту. К ингибиторам TNF относятся:
Врачи могут вводить ингибиторы TNF путем инъекций под кожу, обычно в бедро или живот, или в виде инфузии непосредственно в вену.
Люди принимают эти лекарства в течение длительного времени, и обычно им требуется около 3 месяцев, чтобы заметить разницу.
Как и все лекарства, анти-TNF может вызывать побочные эффекты. К ним относятся боль или припухлость в месте инъекции и повышенный риск инфекций, включая туберкулез и грибковую инфекцию.
Врачи обычно наблюдают за людьми, принимающими препараты против TNF, на предмет признаков побочных эффектов.
Куркумин
Некоторые исследователи предположили, что куркумин, ключевое соединение куркумы, может снизить уровень TNF в крови.
Авторы обзорного исследования, опубликованного в 2013 году, рассмотрели все доступные доказательства связи куркумина с TNF и другими воспалительными маркерами.
Исследователи пришли к выводу, что куркумин действительно способен подавлять пути, ведущие к воспалению. Однако в большинстве исследований, включенных в обзор, использовалась чашка Петри в лаборатории, а не на людях.
Следовательно, исследователи должны провести больше исследований, прежде чем они смогут подтвердить эффективность куркумина как блокатора TNF у людей.
Гранат
Некоторые анекдотические источники предполагают, что экстракты плодов граната могут помочь снизить уровень TNF у людей, живущих с воспалительными заболеваниями.
Однако исследование, опубликованное в 2012 году, не нашло никаких доказательств этого.
TNF — это белок, который способствует воспалению. У здоровых людей это важная часть иммунной системы, помогающая организму противостоять вторжению бактерий и вирусов и излечивать поврежденные ткани.
У людей с аутоиммунными заболеваниями избыточный уровень TNF в крови может привести к ненужному воспалению.Эта реакция может вызвать симптомы, которые часто бывают болезненными.
Врачи прописывают долгосрочные препараты против TNF для лечения многих воспалительных состояний, включая различные формы артрита и ВЗК. Эти методы лечения работают, блокируя активность избыточного TNF в крови.
Факторы некроза опухоли
| Семейство TNF (фактор некроза опухоли) | |
|---|---|
| [[Изображение: Модель TNF-альфа, производимая M. musculus , основана на структуре PDB 2TNF (1.Разрешение 4Å). Разные цвета представляют разные мономеры. Baeyens, KJ et al. (1999). [1] Сгенерировано в FirstGlance Jmol. | 220px]] | |
| Идентификаторы | |
| Символ | TNF |
| Pfam | PF00229 |
| InterPro | IPR006052 |
| ПРОЗИТ | PDOC00224 |
| SCOP | 1тнф |
Факторы некроза опухоли (или TNF-семейство ) относится к группе семейства цитокинов, которые могут вызывать гибель клеток (апоптоз).Первые два члена семьи, которые будут идентифицированы, были:
- Фактор некроза опухоли альфа (TNF-α) является наиболее известным представителем этого класса, и его иногда называют, когда используется термин «фактор некроза опухоли». TNF-α представляет собой цитотоксин, происходящий из моноцитов, который участвует в регрессии опухоли, септическом шоке и кахексии. [2] [3] Белок синтезируется как прогормон с необычно длинной и атипичной сигнальной последовательностью, которая отсутствует в зрелом секретируемом цитокине. [4] Короткий гидрофобный участок аминокислот служит для закрепления прогормона в липидных бислоях. [5] И зрелый белок, и частично процессированная форма гормона могут секретироваться после расщепления пропептида. [5]
- Фактор некроза опухоли-бета (TNF-β), также известный как лимфотоксин, представляет собой цитокин, который ингибируется интерлейкином 10 [6]
Члены семьи
Девятнадцать цитокинов были идентифицированы как часть семейства TNF на основе сходства последовательностей, функций и структуры. [7] Сюда входят: [8] [9] [10]
- Фактор некроза опухоли (TNF) (также известный как кахектин или TNF-альфа) [11] [12] — это цитокин, который выполняет широкий спектр функций. Это может вызвать цитолиз определенных линий опухолевых клеток; участвует в индукции кахексии; это мощный пироген, вызывающий лихорадку прямым действием или стимуляцией секреции интерлейкина-1; он может стимулировать пролиферацию клеток и индуцировать дифференцировку клеток при определенных условиях.
- Лимфотоксин-альфа (LT-альфа) и лимфотоксин-бета (LT-бета), два родственных цитокина, продуцируемых лимфоцитами, которые являются цитотоксичными для широкого спектра опухолевых клеток in vitro и in vivo. [13]
- Т-клеточный антиген gp39 (CD40L), цитокин, который, по-видимому, важен для развития и активации В-клеток.
- CD27L, цитокин, который играет роль в активации Т-клеток. Он индуцирует пролиферацию костимулированных Т-клеток и усиливает образование цитолитических Т-клеток.
- CD30L, цитокин, индуцирующий пролиферацию Т-клеток.
- FASL, цитокин, участвующий в гибели клеток. [14]
- 4-1BBL, индуцируемая поверхностная молекула Т-клеток, которая способствует стимуляции Т-клеток.
- OX40L, цитокин, который стимулирует пролиферацию Т-клеток и выработку цитокинов. [15]
- Связанный с TNF лиганд, индуцирующий апоптоз (TRAIL), цитокин, индуцирующий апоптоз. [16]
Все эти цитокины, по-видимому, образуют гомотримерные (или гетеротримерные в случае LT-альфа / бета) комплексы, которые распознаются их специфическими рецепторами.Сильные водородные связи между мономерами стабилизируют третичную структуру. Одним из таких примеров является водородная связь Asn34-Arg82 в M. musculus TNF alpha [17] .
Модель водородной связи между Asn34 субъединицы A и Arg82 субъединицы C, продуцируемая M. musculus , на основе структуры PDB 2TNF. Остатки, участвующие в водородной связи, показаны карандашом. Короткая длина связи, 2,84 Å, свидетельствует о сильной водородной связи, которая поддерживает третичную структуру.Baeyens, KJ et al. (1999) [18] . Создано в Chimera.Паттерн PROSITE для этого семейства расположен в бета-цепи в центральной части белка, который является консервативным для всех членов.
Все члены семейства TNF, за исключением секретируемого лимфотоксина и лиганда, индуцирующего пролиферацию (APRIL), являются трансмембранными белками типа II, которые выступают из иммунных клеток. Такие связанные с мембраной лиганды TNF часто передают обратный сигнал иммунным клеткам, когда они контактируют и связываются со своими родственными рецепторами на других клетках. Baeyens, KJ et al. (1999). «Структура фактора некроза опухолей мыши с разрешением 1,4 Å в сторону модуляции его селективности и тримеризации». Кристаллография Acta Раздел D: Биологический кристаллографил 55 (Pt4): 772-8. PMID 10089307.
Внешние ссылки
| |||||||||||||||||||||||||||||||||||||||||||
Эта статья включает текст из общественного достояния Pfam и InterPro IPR006052
Frontiers | Фактор некроза опухоли α и регуляторные Т-клетки в онкоиммунологии
TNFR2 на регуляторных Т-клетках (Treg): современное состояние
Иммуносупрессивная функция фактора некроза опухоли α (TNF)
Фактор некроза опухоли α представляет собой плейотропный цитокин, продуцируемый различными типами клеток и участвующий в широком диапазоне патологических процессов [для обзора см. Ссылки (1, 2)].Первоначально его считали провоспалительной молекулой. Однако доклинические и клинические данные показали, что он также опосредует парадоксальный противовоспалительный и иммуномодулирующий эффект. Действительно, на мышиных моделях диабета 1 типа или волчаночного нефрита TNF может оказывать защитное действие (3–7). Более того, у пациентов, получавших терапию анти-TNF, наблюдали новое начало или обострение хронических воспалительных и аутоиммунных заболеваний (8–14). Ниже мы подробно опишем случай болезни «трансплантат против хозяина» (GVHD) как пример амбивалентного действия TNF в иммунопатологии.
Различные возможные механизмы подавления действия TNF
Фактор некроза опухоли α связывается с двумя рецепторами, а именно с рецептором TNF типа 1 (TNFR1) и TNFR2 (рис. 1). В отличие от TNFR1, который имеет повсеместную экспрессию, TNFR2 экспрессируется некоторыми иммунными клетками, предпочтительно фракцией Treg, некоторыми эндотелиальными клетками и клетками нервной ткани (2, 15). Было предложено несколько механизмов для объяснения подавляющего действия TNF. Было показано, что хроническая стимуляция TNF может инактивировать передачу сигналов TCR (16) или вызвать истощение T-клеток (17).В качестве альтернативы цитокин может убивать CD8 + Т-клетки, что особенно важно для аутореактивных клеток (18). Помимо этих присущих клеткам механизмов, TNF может проявлять свою супрессивную активность, стимулируя клетки, обладающие иммуносупрессивным действием, такие как клетки-супрессоры миелоидного происхождения (MDSC) (19, 20). Наконец, пионерские работы Чена и Оппенгейма предположили, что этот иммуносупрессивный эффект TNF может быть связан с прямой активацией Treg (15, 21). Этот последний механизм, который является наиболее изученным и подтверждается данными, полученными различными группами, подробно описан ниже.Обычно считается, что подавляющее действие TNF опосредовано его взаимодействием с TNFR2, поскольку передача сигналов TNFR2, по-видимому, является защитной при различных иммунопатологиях, а некоторые из механизмов, описанных ранее, зависят от TNFR2 (22). Однако, в то время как взаимодействие TNF / TNFR1 в основном описано как провоспалительное, запуск TNFR1 может также подавлять экспрессию IL-12 / IL-23 p40 макрофагами (23). Этот механизм может объяснить парадоксальную экспансию клеток Th2 / Th27 после лечения анти-TNF у пациентов с аутоиммунными заболеваниями, которые не реагируют на эту терапию (24, 25).
Рисунок 1 . Иммунодепрессивное действие фактора некроза опухоли α (TNF). TNF может проявлять свою иммуносупрессивную активность за счет внутреннего негативного воздействия на активацию обычных Т-клеток (Tconvs) или путем усиления супрессивных клеток, таких как миелоидные супрессорные клетки (MDSC) или регуляторные T-клетки (Tregs). На Tconvs долгосрочное действие TNF может способствовать убийству, истощению или инактивации TCR. На MDSC TNF может повышать их активность, способствуя их выживанию, локальному рекрутированию или подавляющей функции.На Tregs TNF может способствовать их пролиферации, выживанию и стабильности.
Экспрессия TNFR2 с помощью Treg
ЭкспрессияTNFR2 повышается в активированных Treg и может быть обнаружена в активированных обычных T-клетках (Tconv), хотя и на более низких уровнях, чем в активированных Treg. Некоторые другие члены семейства TNFR, такие как GITR, OX40 или 4-1BB, также предпочтительно экспрессируются Treg, и их экспрессия также повышается при активации (26). Примечательно, что в транскриптомном анализе при сравнении Treg и Tconv лимфоидных тканей TNFR2, OX40 и GITR принадлежат к сигнатуре Treg, и их экспрессия коррелирует с низким метилированием ДНК в Treg, что позволяет предположить, что их транскрипция, по крайней мере, частично регулируется на эпигенетическом уровне (27 , 28).Эти три молекулы экспрессируются на ранних стадиях линии Treg, начиная со стадии предшественника Treg тимуса, и их экспрессия важна для развития Treg (29). В лимфоидной ткани мышей или в крови человека TNFR2 экспрессируется фракцией активированных Treg, экспрессирующих высокие уровни других маркеров активации, таких как CTLA-4 (30). Экспрессия TNFR2 замечательно идентифицирует подмножество Treg с самой высокой подавляющей способностью (21, 30, 31).
Стимулирующее действие TNF на Tregs через TNFR2
Прямое действие TNF на TNFR2-экспрессирующие Treg было изучено Ченом и Оппенгеймом in vitro и было рассмотрено в другом месте (32).Вкратце, TNF увеличивает пролиферацию, выживаемость, стабильность, экспрессию CD25, Foxp3 и маркеров активации, а также супрессивную функцию Treg мыши (15, 26, 30, 31). Многие из этих эффектов TNF, особенно на пролиферацию, могут быть воспроизведены с человеческими Tregs (32–35). Однако некоторые исследования утверждают, что TNF ингибирует супрессивную активность человеческих Treg (36–39). Интерпретация некоторых из этих исследований была осложнена тем фактом, что TNF может сделать Tconv более устойчивыми к Treg-опосредованному подавлению.После обширного и тщательного изучения этого вопроса мы могли сделать вывод, что TNF не ингибирует супрессивную активность человеческих Treg (35).
Роль TNFR2 в биологии Treg In vivo
Роль in vivo TNFR2 в биологии Treg было труднее оценить из-за отсутствия условного нокаута TNFR2 в Treg. Однако есть убедительные доказательства того, что TNF может увеличивать экспансию Treg при различных воспалительных процессах (40). Мы показали, что TNF, вероятно, продуцируемый Tconvs, стимулировал пролиферацию Treg при диабете 1 типа (41).Другие наблюдали подобное явление во время септического шока, инфекционного заболевания или иммунного ответа (15, 42, 43). Кроме того, TNFR2-дефицитные Treg утратили способность контролировать колит, что было связано с уменьшением выживаемости и стабильности по сравнению с контрольными Treg дикого типа (31, 44). Критическая роль TNFR2, экспрессируемого Treg, также изучалась в контексте РТПХ и рака и будет конкретно обсуждаться ниже. В целом, среди всех эффектов TNF на биологию Treg, его способность увеличивать пролиферацию является наиболее убедительной, поскольку о нем сообщалось во многих исследованиях in vitro, и in vivo, , проведенных различными группами с использованием Treg мыши и человека.Доказательства того, что этот цитокин также увеличивает выживаемость и стабильность Treg, весьма убедительны, и его влияние на функцию Treg требует дальнейшего изучения.
Надежды и разочарование в отношении воздействия на TNF при РТПХ
TNF и TNFR1 как прогностические биомаркеры при РТПХ
Фактор некроза опухоли α играет ключевую роль в острой GVHD (aGVHD), системном и сильно воспалительном осложнении, которое возникает после аллогенной трансплантации гемопоэтических стволовых клеток (allo-SCT) (45). TNF действительно играет важную роль на разных этапах этого патологического процесса, в котором донорские Т-клетки распознают здоровые ткани чужеродного хозяина и в конечном итоге вызывают их разрушение (рис. 2).В этом направлении клинические исследования четко продемонстрировали положительную корреляцию между уровнями растворимого TNFR1, измеренными через 7 дней после трансплантации, и временем до начала и тяжестью оРТПХ (46, 47). Повышение уровней TNFR1 между исходным уровнем и 7-м днем было не только независимым предиктором рТПХ, но также связанной с трансплантацией смертности и общей выживаемости. Кроме того, повышение TNF, измеряемое уровнями белка в периферической крови, уровнями транскрипции РНК или проточной цитометрией, предшествует началу оРТПХ, прежде чем достигнет пика во время его развития (48-50).В целом результаты этих клинических исследований привели к интеграции TNFR1 как части панели биомаркеров, которая может различать пациентов с рТПХ и без них и прогнозировать выживаемость (51).
Рисунок 2 . Надежды и разочарование в отношении воздействия на фактор некроза опухоли α (TNF) при болезни трансплантат против хозяина (GVHD). Лечение анти-TNF способно блокировать действие TNF на различных этапах патофизиологии острой GVHD, включая начальную активацию APC хозяина (1), привлечение и активацию эффекторных Т-клеток в тканях-мишенях (2) и прямой некроз клеток (3).За счет ингибирования связывания TNF с TNFR2, экспрессируемым регуляторными Т-клетками (Tregs), лечение анти-TNF также может оказывать вредное воздействие на эти супрессивные клетки, приводя к усиленному размножению и активации аллореактивных донорских Т-клеток, которые могут быть ответственны за неутешительные результаты. наблюдается при лечении анти-TNF в этой обстановке. Сокращения: APC — антигенпрезентирующая клетка; ЛПС, липополисахарид.
Клинические испытания анти-TNF при РТПХ
Ключевая роль TNF в патофизиологии aGVHD логически побудила исследователей и врачей попытаться заблокировать этот цитокин, чтобы уменьшить воспаление и, следовательно, предотвратить или лечить aGVHD.В этом направлении большая часть клинических испытаний была сосредоточена на двух молекулах: инфликсимабе — моноклональном антителе (mAb), связывающем TNF, и этанерцепте — растворимом TNFR, который конкурирует с клеточными рецепторами за связывание TNF. Большие надежды, возрожденные нацеливанием TNF на рТПХ в течение первого десятилетия этого столетия, к сожалению, быстро исчезли из-за несколько разочаровывающих результатов клинических исследований. Действительно, клинические испытания не подтвердили каких-либо преимуществ от добавления инфликсимаба к стандартному лечению как для профилактики, так и для лечения РТПХ (52, 53).Лишь небольшие ретроспективные исследования показали многообещающую частоту ответа на лечение стероидорезистентной aGVHD, в основном в случае поражения кишечного тракта (54–57). Однако эффективность инфликсимаба при стероидорезистентной рТПХ, по-видимому, не превосходит эффект, наблюдаемый при применении других доступных препаратов (58), хотя проспективные рандомизированные исследования отсутствуют. Более того, другие исследования показали, что реакция после терапии инфликсимабом является плохо устойчивой и вызывает обеспокоенность по поводу повышенного риска тяжелых инфекций (59, 60).
Что касается этанерцепта, одноцентровое проспективное исследование показало многообещающую частоту ответа при комбинации этанерцепта со стандартными высокими дозами кортикостероидов для лечения рТПХ первой линии по сравнению с когортой одновременных сопоставимых пациентов, получавших только высокие дозы кортикостероидов (61). . Однако более высокая частота ответа, наблюдаемая при применении этанерцепта, не приводила к значительному увеличению выживаемости через 6 месяцев от начала рТПХ. Более того, многоцентровое проспективное рандомизированное исследование «выберите победителя», сравнивающее четыре многообещающих молекулы в комбинации с кортикостероидами для лечения оРТПХ первой линии, выявило микофенолятмофетил, а не этанерцепт, как наиболее многообещающий агент (62).Однако микофенолятмофетил не продемонстрировал каких-либо преимуществ в последующем многоцентровом рандомизированном двойном слепом плацебо-контролируемом исследовании фазы 3, в котором оценивали его добавление к стандартным кортикостероидам (63). В условиях стероидорезистентной оРТПХ два небольших одноцентровых исследования показали лишь умеренный эффект этанерцепта с несколькими полными ответами (64, 65). Что касается инфликсимаба, его эффективность была выше при поражении кишечника. Наконец, в исследовании фазы 2 с участием 100 пациентов также оценивали этанерцепт как часть профилактики оРТПХ в сочетании с такролимусом и низкими дозами метотрексата (66).И снова польза этанерцепта не была очевидной, поскольку его добавление к стандартной профилактике не повлияло на общий риск aGVHD 2–4 степени по сравнению с контрольной группой из 161 ранее зарегистрированного пациента. В этом исследовании предполагалась только потенциальная польза для пациентов, не облученных полностью. Мониторинг уровня TNFR1 в плазме также может использоваться для оценки и / или прогнозирования ответа на лечение этанерцептом, поскольку у пациентов, ответивших на лечение, наблюдалось значительное снижение этих уровней (61, 66).Подводя итог, можно сказать, что нынешнее место лечения анти-TNF в арсенале aGVHD ограничивается только терапевтическим вариантом для стероидорезистентного заболевания, в основном в случае поражения кишечного тракта. Возможное объяснение этой неудачи состоит в том, что блокирование TNF также будет влиять на TNFR2-зависимое повышение Treg, которое является защитным при GVHD, как предполагают экспериментальные данные, обсуждаемые ниже.
Надежда на борьбу с TNFR2 (и Tregs) при РТПХ
Регуляторные Т-клетки модулируют аллореактивность во время алло-SCT.Клеточная терапия с использованием Treg эффективно контролирует GVHD (67, 68), тогда как делеция Treg может использоваться для усиления эффекта трансплантат против лейкемии (GVL) (69). Таким образом, некоторые исследовательские группы рассматривали TNFR2 как потенциальную мишень для воздействия непосредственно на Treg в этих условиях и модуляции аллореактивности с агонистами или антагонистами TNFR2. В этой связи в 2016 г. почти одновременно были опубликованы три важных исследования (70–72).
На мышиной модели профилактики aGVHD, основанной на инфузии Treg, мы ясно показали с помощью трех различных экспериментальных подходов, что защитный эффект, опосредованный терапевтическими Treg, зависит от TNF, продуцируемого патогенным Tconv, и TNFR2, экспрессируемого Treg (71).Действительно, при блокировании взаимодействия TNF / TNFR2 с mAb против TNFR2 или при использовании eith
Frontiers | Фактор некроза опухоли, связанный с рецептором, регуляция ядерного фактора κB и пути митоген-активируемых протеинкиназ
Введение
Факторы, ассоциированные с рецептором фактора некроза опухоли (TNFR) (TRAF), образуют семейство внутриклеточных сигнальных молекул, которое в клетках млекопитающих включает шесть типичных членов (TRAF1-6) и атипичного члена (TRAF7) (1, 2).Типичные члены TRAF имеют сходную вторичную структуру, включая гомологичный С-концевой домен, называемый доменом TRAF, и различное количество цинковых пальцев. Кроме того, все члены TRAF, кроме TRAF1, содержат RING-домен, расположенный в N-концевой области. Домен TRAF опосредует олигомеризацию белков TRAF, а также их ассоциацию с вышестоящими рецепторами или адаптерами и последующими эффекторными белками (1). Домен RING наиболее известен своей функцией опосредовать убиквитинирование белка в большом семействе лигаз убиквитиназы E3 (3).TRAF6 представляет собой хорошо охарактеризованную лигазу E3, которая специфически конъюгирует лизин (K) 63-связанные цепи полиубиквитина (4). Было показано, что несколько других белков TRAF, TRAF2, TRAF3 и TRAF5 обладают K63-специфическими функциями E3, хотя физиологическая функция их активности E3 менее четко определена (1, 2, 5).
Первоначально идентифицированные как сигнальные адаптеры TNFR2 (6), теперь известно, что молекулы TRAF опосредуют передачу сигнала от большого количества иммунных рецепторов, включая членов суперсемейства TNFR и другие рецепторы цитокинов, рецепторы распознавания образов (PRR) и рецепторы антигенов. (1, 2).Среди основных нижестоящих путей, регулируемых TRAF, находятся пути, ведущие к активации ядерного фактора κB (NF-κB) и митоген-активируемых протеинкиназ (MAPK), которые, в свою очередь, важны для индукции генов, связанных с врожденным иммунитетом, воспалением и выживаемость клеток (7, 8). В дополнение к этим классическим функциям в недавних исследованиях были обнаружены новые функции TRAF. В частности, некоторые белки TRAF, включая TRAF2 и TRAF3, действуют как негативные регуляторы в некоторых сигнальных путях, участвующих в выживании В-клеток и воспалительных ответах клеток врожденного иммунитета (9).В этом обзоре основное внимание уделяется роли TRAFs в регуляции сигнальных путей NF-κB и MAPK.
Пути NF-κB и MAPK
Дорожки NF-κB
Ядерный фактор κB — это семейство индуцибельных факторов транскрипции, которые регулируют множество биологических процессов, включая иммунные и воспалительные реакции, рост и выживание клеток, а также онкогенез (8, 10, 11). Семейство NF-κB млекопитающих включает пять структурно родственных членов, RelA, RelB, c-Rel, NF-κB1 p50 и NF-κB2 p52, которые связываются с консервативным элементом ДНК (κB) в виде различных гомо- и гетеродимеров в промоторные или энхансерные области генов-мишеней (10).NF-κB обычно изолируются в цитоплазме в виде неактивных комплексов, связанных семейством ингибиторных белков, ингибиторных κB (IκB), которое включает IκBα, IκBβ, и несколько других структурно родственных белков, характеризующихся присутствием домена анкирин-повтора, опосредующего связывание. и ингибирование NF-κBs (10). NF-κB1 и NF-κB2 продуцируются как белки-предшественники, p105 и p100, оба из которых содержат IκB-подобную C-концевую часть и функционируют как IκB-подобные молекулы (12, 13). Эти белки-предшественники могут процессироваться протеасомой, что включает селективную деградацию IκB-подобной C-концевой части, тем самым продуцируя зрелые NF-κB p50 и p52 соответственно, и нарушая их IκB-подобную функцию.Во время его трансляции около половины количества p105 постоянно процессируется для продукции p50, тогда как оставшийся p105 функционирует как IκB, чтобы регулировать ядерную транслокацию различных членов NF-κB, включая p50, RelA и c-Rel (13–16). В отличие от обработки p105, обработка p100 строго контролируется и происходит индуцируемым сигналом образом (17).
Два основных пути передачи сигналов, канонический и неканонический пути, обеспечивают активацию членов NF-κB (18).Канонический путь основан на индуцированной сигналом деградации IκB, особенно IκBα, которая запускает ядерную транслокацию p50-содержащих комплексов NF-κB, в частности гетеродимеров p50 / RelA и p50 / c-Rel. Этот путь основан на активации комплекса тримерной киназы IκB (IKK), состоящего из двух каталитических субъединиц, IKKα и IKKβ, и регуляторной субъединицы, эссенциального модулятора NF-κB (NEMO) (также называемого IKKγ). Активация IKK обычно опосредуется трансформирующей бета-активированной киназой 1 (TAK1) фактора роста, киназой киназы MAPK (MAP3K), которая реагирует на различные сигналы иммунных рецепторов и полагается на убиквитинирование для своей каталитической активации и сигнальной функции (19, 20).Дефицит TAK1 сильно ослабляет, хотя и не полностью блокирует индуцированную IL-1 и TNFα активацию NF-κB (21). Другой MAP3K, MEKK3, также участвует в активации NF-κB с помощью различных индукторов, таких как TNFα и IL-1 (22–27). Характеристикой канонического сигнального пути NF-κB является его быстрая и временная кинетика, которая важна для предотвращения дерегулируемых функций NF-κB. Канонический путь NF-κB играет важную роль в регуляции различных иммунных функций, включая врожденный иммунитет и воспаление, активацию и дифференцировку лимфоцитов, а также иммунную толерантность (8, 28).
Неканонический путь NF-κB основан на процессинге белка-предшественника NF-κB2 p100, который запускается посредством сайт-специфического фосфорилирования в его C-концевых остатках серина (17). Этот путь не зависит от TAK1 и тримерного комплекса IKK, но требует NF-κB-индуцирующей киназы (NIK) и ее нижестоящей киназы IKKα (17, 18, 29). В отличие от канонического пути NF-κB, который отвечает на сигналы от большого количества иммунных рецепторов, неканонический путь NF-κB избирательно реагирует на подмножество членов надсемейства TNFR, включая CD40, рецептор BAFF, рецептор лимфотоксина β (LTβR), RANK , TNFR2, TWEAK, CD27 и др.(30, 31). Отличительной чертой неканонической передачи сигналов NF-κB является его медленная кинетика и зависимость от синтеза белка (31). Во многом это связано с участием необычного механизма регуляции NIK. В нормальных условиях стабильный уровень NIK чрезвычайно низок из-за его конститутивной деградации через убиквитин / протеасомный путь, что предотвращает индукцию процессинга p100 (32). Стимулируемая сигналом неканоническая передача сигналов NF-κB включает стабилизацию синтезированного de novo NIK, тем самым позволяя накопленным NIK активировать IKKα и индуцировать процессинг p100.Неканонический путь NF-κB наиболее известен своей ролью в регуляции развития лимфоидных органов, созревания В-клеток и дифференцировки остеокластов. Однако недавние исследования выявили дополнительные функции этого пути и связали этот путь с аутоиммунными и воспалительными заболеваниями (18).
Пути MAPK
Активированные митогеном протеинкиназы образуют большое семейство серин / треониновых киназ, которые отвечают на разнообразные внеклеточные и внутриклеточные стимулы и опосредуют множество биологических процессов, таких как рост и дифференцировка клеток, иммунные и воспалительные реакции и онкогенез (7, 33).Семейство MAPK млекопитающих включает три подсемейства: киназы, регулируемые внеклеточными сигналами (ERK), N-концевые киназы c-Jun (JNK) и p38 MAPK (7, 33). Сигнальная трансдукция MAPKs имеет общий механизм, в котором MAPK фосфорилируется и активируется киназой MAPK (MKK), а MKK, в свою очередь, фосфорилируется и активируется MAP3K. Различные MAPK регулируются разными MKK: MKK1 и MKK2 (также называемые MEK1 и MEK2) для ERK1 и ERK2, MKK4 и MKK7 для JNK и MKK3 и MKK6 для p38.MAP3K для сигнальных каскадов MAPK различаются для разных стимулирующих рецепторов. В иммунной системе сигнальные каскады MAPK были тщательно изучены на клетках врожденного иммунитета, стимулированных через PRR, в частности толл-подобных рецепторов (TLR), где MAPK опосредуют индукцию различных провоспалительных цитокинов и хемокинов, необходимых для защиты хозяина и воспаления ( 7).
В пути TLR TAK1 является первичным MAP3K, опосредующим активацию сигнальных каскадов JNK и p38, тогда как Tpl2 является MAP3K, опосредующим активацию каскада ERK1 / 2 (34).Передача сигналов MAPK, стимулированная TLR, включает интересные перекрестные помехи с путем IKK / NF-κB. Белок-предшественник NF-κB1 p105 образует стабильный комплекс с MAP3K Tpl2, в котором p105 одновременно стабилизирует Tpl2 и предотвращает его фосфорилирование нижележащих киназ, MKK1 / 2 (35–39). После активации сигналом TLR IKK фосфорилирует p105, чтобы вызвать его убиквитин-зависимую деградацию, что позволяет освобожденному Tpl2 фосфорилировать MKK1 / 2, что приводит к активации ERK1 / 2. Это сигнальное событие является временным, поскольку активированный Tpl2 быстро деградирует из-за его нестабильности при диссоциации от p105.Следовательно, в пути TLR TAK1 функционирует как основная киназа, обеспечивающая активацию не только каскадов JNK и p38 MAPK, но также и оси передачи сигналов IKK-Tpl2-ERK.
TRAF6 как медиатор активации NF-κB и MAPK
Активация IKK / MAPK
TRAF6 является прототипом убиквитинлигазы E3, содержащей RING-домен, которая функционирует вместе с димером E2, состоящим из Ubc13 и Uev1A, специфически катализируя синтез K63-связанных цепей полиубиквитина (4, 19).Накапливающиеся исследования продемонстрировали решающую роль TRAF6 в передаче сигналов от различных TNFR, а также других иммунных рецепторов, таких как рецептор IL-1 (IL-1R), IL-17R, TLR, RIG-I-подобные рецепторы, TGFβR, и рецепторы антигенов (20, 40–44). С-концевой домен TRAF TRAF6 взаимодействует с консервативным мотивом последовательности Pro-X-Glu-XX- (ароматический / кислотный остаток), присутствующим у определенных членов суперсемейства TNFR, включая CD40 и активатор рецептора ядерного фактора каппа-B. (45). Посредством этого молекулярного взаимодействия TRAF6 рекрутируется на TNFR в ответ на стимуляцию лигандом, что важно для запуска активации TRAF6 и передачи сигнала.TRAF6-связывающие мотивы также присутствуют в сигнальных адаптерах других иммунных рецепторов, таких как IL-1R-ассоциированные киназы (IRAK) путей IL-1R и TLR, митохондриальный антивирусный сигнальный белок пути RIG-I и Act1 пути IL-17R (41, 44, 46).
Сигнальный механизм TRAF6 широко изучен в путях TLR и IL-1R (Рисунок 1). TLR передают сигналы через общие адаптеры , MyD88 и TRIF (47). За исключением TLR3, который передает сигнал через TRIF, все TLR, а также IL-1R полагаются на MyD88 для передачи сигнала, хотя TLR4 передает сигналы через MyD88 и TRIF.При связывании лиганда MyD88-зависимые TLR рекрутируют IRAK (включая IRAK1, IRAK2 и IRAK4) через адаптер MyD88 , чтобы запустить образование связанного с рецептором сигнального комплекса, который также содержит TRAF6 (Рисунок 1). После активации в сигнальном комплексе MyD88, TRAF6 функционирует как убиквитинлигаза E3, которая конъюгирует K63-связанные цепи убиквитина с собой, а также с другими белками, такими как IRAK1 и регуляторная субъединица IKK, NEMO (19, 20). Не совсем понятно, как именно TRAF6 обеспечивает активацию нижестоящих путей.Обычно считается, что самоубиквитинированный TRAF6 рекрутирует нижестоящие киназы TAK1 и IKK для сборки сигнального комплекса, который способствует активации TAK1 и IKK. В подтверждение этой модели и TAK1, и IKK содержат регуляторные субъединицы, которые связывают K63-связанные цепи полиубиквитина (48-50). Комплекс TAK1 содержит две регуляторные субъединицы, TAB 1 и TAB 2 (или TAB 3), а TAB 2 и TAB 3 оба содержат убиквитин-связывающий домен типа Npl4 Zinc Finger, который специфически связывает K63-связанные цепи полиубиквитина (48, 51) .Регуляторная субъединица IKK, NEMO, также содержит убиквитин-связывающий домен, называемый CC2-LZ (coiled-coil2-lucine zipper) или UBAN (связывание убиквитина в белках ABIN и NEMO), способный связывать цепи убиквитина K63 (49, 50 , 52–54). В качестве дополнительного подтверждения этой модели мутация сайта автоубиквитинирования, K124, TRAF6 ослабляет его функцию в обеспечении активации TAK1 / IKK (55). Однако последующее исследование предполагает, что E3-лигазная активность TRAF6, хотя и необходима для активации TAK1, не обязательна для ассоциации TRAF6 с комплексом TAK1 (56).Более того, это исследование также предполагает, что аутоубиквитинирование TRAF6 необходимо для активации TAK1 и его нижестоящих путей NF-κB и MAPK с помощью IL-1 и RANKL, тогда как TRAF6-опосредованное убиквитинирование NEMO способствует активации IKK и NF-κB (56) . Поскольку неконъюгированные цепи убиквитина способны активировать TAK1 (57), также вероятно, что как конъюгированные, так и неконъюгированные цепи убиквитина K63 в комплексе TRAF6 / TAK1 вносят вклад в активацию TAK1.
Рисунок 1 .Функция и регуляция фактора, ассоциированного с рецептором фактора некроза опухоли (TRAF) 6 в сигнальном пути MyD88. При стимуляции лигандами IL-1 или toll-подобного рецептора (TLR) MyD88 рекрутирует связанные с IL-1R киназы (IRAK) (включая IRAK1, IRAK2 и IRAK4) и TRAF6 для сборки сигнального комплекса MyD88. После активации в комплексе MyD88 TRAF6 действует как убиквитин-лигаза E3, которая катализирует синтез K63-связанных полиубиквитиновых цепей, конъюгированных с самим собой или основным модулятором NF-κB (NEMO), или существующих в виде свободных цепей убиквитина.Самоубиквитинированный TRAF6 рекрутирует убиквитин-зависимую киназу, трансформирующую фактор роста, бета-активированную киназу 1 (TAK1) и ее нижележащую киназу IκB (IKK) для сборки сигнального комплекса, который способствует активации TAK1 и IKK. Для этого процесса необходима регуляторная субъединица TAK1 TAB 2 (или TAB 3) и регуляторная субъединица IKK NEMO, обе обладают убиквитин-связывающими функциями. Активированный TAK1 опосредует активацию IKK и митоген-активированных протеинкиназ (MAPK), которые дополнительно активируют ядерный фактор κB (NF-κB) (RelA- и c-Rel-содержащие комплексы) и AP1.TRAF6-опосредованное убиквитинирование NEMO также способствует активации IKK и NF-κB. Было показано, что несколько DUB негативно регулируют функцию TRAF6 посредством деконъюгации его полиубиквитиновых цепей K63.
Хотя активность E3 ubiquitin ligase TRAF6 обычно считается существенной для его сигнальной функции, существуют разногласия. Недавнее исследование, в котором использовались методы восстановления клеточной линии и мышиный нокин, демонстрирует, что инактивация E3-лигазной активности TRAF6 лишь частично подавляет его функцию в опосредовании активации передачи сигналов NF-κB и MAPK, стимулированной IL-1, TLR и RANKL (58). .Похоже, что семейство Pellino лигаз E3, особенно Pellino1 и Pellino2, может компенсировать функцию E3 TRAF6, чтобы опосредовать конъюгацию цепи убиквитина K63 для активации TAK1 (58). Поскольку полная делеция TRAF6 отменяет MyD88-зависимую активацию передачи сигналов NF-κB и MAPK, эти находки предполагают, что помимо того, что он служит лигазой E3, TRAF6 может также функционировать как адаптер.
Регламент TRAF6
Принимая во внимание решающую роль TRAF6 в опосредовании активации путей NF-κB и MAPK и воспаления, неудивительно, что функция TRAF6 находится под жестким контролем различных регуляторов (Рис. 1).Раннее исследование предполагает, что стимулированное IL-1 полиубиквитинирование TRAF6, индикатор активации TRAF6, происходит временно, что указывает на его регуляцию посредством деубиквитинирования (59). Некоторые DUB участвуют в регуляции активации TRAF6 посредством деконъюгации его полиубиквитиновых цепей K63; к ним относятся CYLD, A20, Otud7b (также называемый Cezanne), USP2a, USP4, USP20, а также myb-подобный SWIRM и домен MPN 1 (60–66). Другой DUB, YOD1 (также называемый Otud2), ингибирует убиквитинирование TRAF6 и сигнальную функцию через некаталитический механизм, который включает связывание с C-концевым доменом TRAF TRAF6 и, таким образом, предотвращение взаимодействия TRAF6 с активирующим адаптерным белком, p62 (также называемый Секестосомой-1) (67).Неясно, почему так много DUB участвует в регуляции TRAF6, но вполне вероятно, что они функционируют в разных типах клеток и / или разных рецепторных путях. Также важно отметить, что роль некоторых TRAF6-регулирующих DUBs в регуляции передачи сигналов TLR и врожденного иммунитета еще предстоит продемонстрировать с использованием подходов in vivo .
РегулированиеTRAF6 также связано с множеством других факторов. Было показано, что член семейства NOD-подобных рецепторов (NLR), NLRC3, ингибирует передачу сигналов от MyD88-зависимых TLR (68).NLRC3 взаимодействует с K63 и ингибирует убиквитинирование TRAF6, тем самым негативно регулируя активацию NF-κB, стимулированную TLR. Интересно, что NLRC3-опосредованная регуляция TRAF6 не влияет на сигнальные пути MAPK (68), указывая на иную роль K63 убиквитинирования TRAF6 в регуляции путей NF-κB и MAPK. Молекулярный механизм, с помощью которого NLRC3 ингибирует убиквитинирование K63 и сигнальную функцию TRAF6, не определен. Предполагаемая молекула E2, Ube2o связывается с TRAF6 и ингибирует убиквитинирование TRAF6 K63 и активацию NF-κB в путях IL-1β и LPS (69).Эта функция Ube2o не зависит от его убиквитин-конъюгированного домена и, по-видимому, действует через нарушение связывания TRAF6 с MyD88. TRAF6 также регулируется протеинкиназой MST4, которая фосфорилирует TRAF6 по двум остаткам треонина (T463 и T486) в C-концевом домене TRAF и ингибирует активность в отношении олигомеризации и автоубиквитинирования TRAF6 (70). Нокдаун MST4 способствует передаче сигналов TLR и индукции цитокинов в культуре клеток и повышает чувствительность мышей к индукции септического шока (70). В отличие от отрицательной роли MST4 в регуляции TRAF6, другая киназа, RSK2, положительно регулирует функцию TRAF6 и активацию MAPK и NF-κB, стимулированную LPS и IL-1β (71).RSK2 фосфорилирует TRAF6 по трем N-концевым серинам (S46, S47 и S48), тем самым стимулируя убиквитинирование K63 и сигнальную функцию TRAF6 (71). В будущих исследованиях следует изучить, фосфорилируются ли эти отрицательные (T463 и T486) и положительные (S46, S47 и S48) сайты фосфорилирования in vivo вместе со стимуляцией TLR.
TRAF2 в сигналах TNFR
TRAF2 широко изучался как медиатор передачи сигналов TNFR1 и, как известно, имеет решающее значение для TNFα-стимулированных путей NF-κB и MAPK.TRAF2 необходим для TNFα-индуцированной активации JNK, хотя он функционально дублирует TRAF5 при активации NF-κB (72, 73). TRAF2 не связывает напрямую TNFR1, но он может рекрутироваться на TNFR1 через адаптерный белок TNF-рецептор-ассоциированный домен смерти (TRADD) (74, 75). Цитоплазматический хвост TNFR1 содержит домен смерти, который опосредует взаимодействие с доменом смерти TRADD, а TRADD связывается с TRAF2 через N-концевую область TRADD и C-концевой TRAF-домен TRAF2 (74, 76).Передача сигналов TNFR1 включает последовательное образование двух различных комплексов, комплекса I и комплекса II, которые опосредуют выживание и гибель клеток соответственно (77) (рис. 2). Комплекс I связан с TNFR1 и состоит из TRADD, TRAF2 и его близкого гомолога TRAF5, рецептор-взаимодействующей протеинкиназы 1 (RIP1) и убиквитинлигаз E3 cIAP1 и cIAP2 (77). В ответ на стимуляцию TNFR1 cIAP1 и cIAP2 конъюгируют K63-связанные цепи убиквитина с RIP1, что способствует привлечению и активации киназных комплексов TAK1 и IKK, что приводит к активации NF-κB и индукции генов выживания (49, 78–80).После начальной передачи сигнала комплекс I диссоциирует от TNFR1, что приводит к образованию цитоплазматического комплекса IIa, состоящего из TRAF2, RIP1, TRADD, FADD и прокаспазы-8. Этот второй комплекс запускает активацию каспазы-8 и апоптоз, который служит контрольным механизмом, опосредующим гибель клеток, когда активация NF-κB, опосредованная комплексом I, не удается (77). TNFR1 также индуцирует комплекс (комплекс IIb), состоящий из RIP1, RIP3 и MLKL, который опосредует гибель клеток через некроптоз (81).
Рисунок 2 .Фактор, связанный с рецептором фактора некроза опухоли (TRAF) 2 и TRAF5 в передаче сигналов рецептора фактора некроза опухоли (TNFR) 1. Связывание TNFα с TNFR1 запускает сборку TNFR1-ассоциированного сигнального комплекса, комплекса I, который состоит из домена смерти, связанного с рецептором TNF (TRADD), рецептор-взаимодействующей протеинкиназы 1 (RIP1), TRAF2 или TRAF5 (TRAF2 / 5) и убиквитин-лигазы E3 cIAP1 и cIAP2 (cIAPs). После активации cIAP конъюгируют K63-связанные цепи убиквитина с RIP1, что способствует привлечению и активации бета-активированной киназы 1 (TAK1) трансформирующего киназу фактора роста, а также привлечению линейного комплекса убиквитин-лигазы LUBAC.LUBAC конъюгирует линейные убиквитиновые цепи с RIP1 и, таким образом, облегчает потребность киназы IκB (IKK) через линейную убиквитин-связывающую функцию эссенциального модулятора NF-κB (NEMO). Последующее убиквитинирование NEMO под действием LUBAC, наряду с TAK1-опосредованным фосфорилированием IKKβ, приводит к активации IKK. Активированные IKK и TAK1 опосредуют активацию ядерного фактора κB (NF-κB) и сигнальных путей митоген-активируемой протеинкиназы (MAPK) / AP1, которые способствуют выживанию клеток. TRAF2 также участвует в последующем образовании цитоплазматического комплекса IIa, который опосредует индукцию апоптоза.Когда каспаза-8 ингибируется, передача сигналов TNFR1 также приводит к образованию комплекса IIb, что приводит к некроптозу. DUB, включая OTULIN и CYLD, негативно регулируют сигнальные функции LUBAC, расщепляя линейные цепи убиквитина. SPATA2, как партнер связывания с высоким сродством CYLD и LUBAC, облегчает функцию CYLD, рекрутируя CYLD в LUBAC.
Подобно TRAF6, TRAF2 содержит N-концевой домен RING и участвует как K63-специфическая убиквитинлигаза E3 (82). Однако структура домена RING TRAF2 значительно отличается от структуры TRAF6, и TRAF2 не взаимодействует с Ubc13 и некоторыми другими E2 (83).Последующее исследование предполагает, что функция TRAF2 лигазы E3 требует кофактора, сфингозин-1-фосфата (S1P), который связывается с доменом RING TRAF2 и стимулирует TRAF2-опосредованное K63 убиквитинирование RIP1 (84). TRAF2 также взаимодействует со сфингозинкиназой 1 (SphK1), одним из изоферментов, катализирующих образование S1P, а siRNA-опосредованное подавление SphK1 ослабляет стимулированное TNFα убиквитинирование RIP1 и активацию NF-κB (84, 85). Вопрос о том, функционирует ли TRAF2 как адаптер или как E3-лигаза в пути TNFR1, все еще обсуждается.В то время как TRAF2 участвует в качестве E3, который опосредует TNFα-индуцированное убиквитинирование RIP1 и активацию NF-κB (84, 86), другие исследования предполагают, что RING-домен TRAF2 является незаменимым для его функции в опосредовании TNFα-индуцированной канонической передачи сигналов NF-κB. (87). Обычно считается, что привлекаемые TRAF2 cIAP действуют как E3-лигазы RIP1 в комплексе I TNFR1 (фиг. 2). CIAP-связывающий мотив, но не RING-домен TRAF2 важен для TNFR1-стимулированного убиквитинирования RIP1 и канонической активации NF-κB (87).cIAP1 и cIAP2 непосредственно убиквитинируют RIP1 in vitro и необходимы для TNFα-индуцированного убиквитинирования RIP1 и активации NF-κB in vivo (78–80).
TRAF2 функционально дублирует TRAF5 в опосредовании активации NF-κB, стимулированной TNFα. Эмбриональные фибробласты мыши (MEF), дефицитные по TRAF2 или TRAF5, полностью функциональны при активации TNF-α-стимулированной NF-κB, тогда как MEF, лишенные обоих членов TRAF, сильно ослаблены, хотя и не полностью дефектны, в активации NF-κB (73).Эти находки подтверждают важную роль TRAF2 и TRAF5 в передаче сигналов NF-κB, стимулированной TNFα, а также предполагают участие дополнительных механизмов. Роль cIAP зависит от типа клетки. shRNA-опосредованный нокдаун cIAP1 в скелетных миобластах в значительной степени блокирует TNFα-стимулированную передачу сигналов NF-κB, тогда как одновременный нокдаун или нокаут cIAP1 и cIAP2 требуется для блокирования активации NF-κB в MEF (79, 88).
Недавние исследования предполагают, что сигнальная функция TNFR1 комплекса I также включает другую убиквитинлигазу E3, LUBAC, которая специфически конъюгирует линейные цепи убиквитина (89, 90).LUBAC — это комплекс, состоящий из трех субъединиц: SHARPIN, HOIL-1L и HOIP. В ответ на стимуляцию TNFR1 LUBAC рекрутируется в комплекс I TNFR1 через связывание с цепями убиквитина K63, конъюгированными с RIP1 с помощью cIAP (89) (рис. 2). Внутри комплекса TNFR1 LUBAC опосредует линейное убиквитинирование нескольких компонентов комплекса 1 TNFR1, включая RIP1 и регуляторную субъединицу IKK NEMO (91, 92). LUBAC-катализируемый синтез линейных цепей убиквитина играет важную роль в привлечении и активации IKK.В отличие от TAB 2 и TAB 3, которые специфически связываются с цепями убиквитина K63, NEMO предпочтительно связывается с линейными цепями убиквитина посредством своего убиквитин-связывающего домена, UBAN (53, 93–95). K63 ubiquitin- и линейные ubiquitin-конъюгированные молекулы RIP1 рекрутируют комплексы TAK1 и IKK, соответственно, тем самым облегчая активацию IKK с помощью TAK1 (Figure 2). Линейное убиквитинирование NEMO может напрямую способствовать активации IKK. Было высказано предположение, что NEMO, конъюгированный с линейной убиквитиновой цепью, может связываться с другой молекулой NEMO, которая способствует димеризации IKK и каталитической активации (96).Также было высказано предположение, что связывание NEMO с линейными цепями убиквитина может вызывать конформационные изменения в IKK, тем самым облегчая фосфорилирование IKKβ с помощью TAK1 (97). Сигнальная функция LUBAC негативно регулируется с помощью DUB, способных расщеплять линейные цепи убиквитина, включая OTULIN и CYLD (98–100). Кроме того, DUB A20 рекрутируется в комплекс TNFR1 посредством связывания с линейными цепями убиквитина и вносит вклад в ингибирование активации NF-κB, возможно, предотвращая рекрутирование NEMO (101, 102).
TRAF2 также является медиатором передачи сигналов TNFR2. Фактически, TRAF2, наряду с TRAF1, был первоначально идентифицирован как сигнальные адаптеры, физически связанные с TNFR2 (6). TRAF2 связывается с цитоплазматическим доменом TNFR2 и рекрутирует E3-убиквитинлигазы cIAP1 и cIAP2 в сигнальный комплекс TNFR2, который важен для активации NF-κB, стимулированной TNFR2 (103, 104). Цитоплазматический хвост TNFR2 содержит мотивы последовательности, которые напрямую связаны с TRAF2 (6, 105, 106). TRAF2 также обеспечивает активацию путей NF-κB и MAPK, запускаемых другими членами суперсемейства TNFR, такими как CD40, OX40, 4-1BB, LTβR и GITR (107–111).
TRAF-контроль неканонического пути NF-κB
Хотя белки TRAF обычно известны как сигнальные адаптеры, которые опосредуют активацию путей NF-κB и MAPK, теперь ясно, что TRAF2 и TRAF3 являются основными негативными регуляторами неканонического пути NF-κB (18). Первоначальное исследование идентифицировало TRAF3 как белок, физически взаимодействующий с неканоническим NIK и опосредующий убиквитин-зависимую деградацию NIK (32). Нокдаун TRAF3 вызывает стабилизацию NIK и индукцию процессинга p100, а индуцированная сигналом неканоническая активация NF-κB связана с деградацией TRAF3 и сопутствующим накоплением NIK, предполагая, что TRAF3-опосредованная деградация NIK является центральным механизмом неканонической регуляции NF-κB ( 32).Это открытие подтверждается последующими исследованиями с использованием мышей с нокаутом TRAF3, демонстрирующими, что дефицит TRAF3 вызывает накопление NIK и неканоническую активацию NF-κB (112–114). Анализ картирования доменов выявил N-концевой мотив NIK, ISIIAQA (аминокислоты 78–84), который необходим для взаимодействия NIK – TRAF3. Мутант NIK, несущий делецию этого мотива, названный NIKΔ78–84 или NIKΔT3, стабилен из-за нарушения взаимодействия с TRAF3 (32). Трансгенная экспрессия этого мутанта NIK в В-клетках вызывает максимальную неканоническую активацию NF-κB и выживание В-клеток независимо от BAFF, что приводит к резкой гиперплазии В-клеток (115).
В отличие от TRAF3, TRAF2 не связывает N-концевую область NIK, а лишь слабо взаимодействует с NIK через C-концевую область (32, 116). Интересно, однако, что дефицит TRAF2 также вызывает неканоническую активацию NF-κB (114, 117). Более того, E3 ubiquitin ligases cIAP1 и cIAP2, как позже было обнаружено, опосредуют убиквитинирование и деградацию NIK (118, 119). Биохимические и генетические данные подтверждают, что TRAF2, TRAF3 и cIAP действуют вместе как комплекс убиквитин-лигазы E3, опосредуя убиквитинирование NIK (120, 121).Было предположено, что внутри этого комплекса TRAF2 и TRAF3 связывают cIAPs и NIK, соответственно, и рекрутируют NIK на лигазы E3 cIAPs посредством димеризации TRAF2 – TRAF3 (31, 120, 121). Эти результаты обеспечивают механистическое понимание функций низкомолекулярных антагонистов IAP при лечении рака. Недавние исследования показывают, что антагонисты IAP не только вызывают гибель раковых клеток, но также способствуют противоопухолевому иммунитету и действуют синергично с ингибиторами иммунных контрольных точек в мышиных моделях иммунотерапии рака (122–125).Иммуностимулирующее действие антагонистов IAP, вероятно, связано с активацией NIK-зависимой неканонической активации NF-κB. Кроме того, антагонисты IAP могут также активировать клетки врожденного иммунитета в микроокружении опухоли. Как будет обсуждаться в следующем разделе, нарушение комплекса TRAF – cIAP E3 в макрофагах способствует продукции провоспалительных цитокинов типа M1, которые облегчают рекрутирование противоопухолевых эффекторных Т-клеток в микроокружение опухоли (126).
Точно, как собирается комплекс TRAF – cIAP и регулирует стабильность NIK, не совсем понятно.Более поздние исследования показывают, что др. Член TRAF, TRAF1, также может участвовать в функции комплекса cIAP E3 и регуляции NIK (110, 127). TRAF1 и TRAF2 образуют гетеротример TRAF1: (TRAF2) 2, который связывает cIAP2 сильнее, чем TRAF2 (127). Поскольку TRAF1 индуцируется различными клеточными стимулами, он может функционировать как модификатор комплекса TRAF-cIAP при определенных условиях. В поддержку этой идеи, TRAF1, по-видимому, играет роль в сдерживании стимулируемой TCR неканонической активации NF-κB (110). Делеция TRAF1 позволяет мышиным Т-клеткам CD8 отвечать на стимуляцию TCR в отсутствие костимуляции 4-1BB для активации неканонического NF-κB (110).Тем не менее, роль TRAF1 в передаче сигналов NF-kB является спорным, так как другое исследование предполагает, что TRAF1 непосредственно связывается NIK и стабилизирует NIK, препятствуя NIK ассоциации с TRAF2-cIAP2 комплекса в условиях избыточной экспрессии (128). Однако важно отметить, что связывание TRAF1-NIK существенно слабее, чем взаимодействие TRAF3-NIK (32), и необходимы дополнительные исследования для изучения физиологической роли TRAF1 в неканонической регуляции NF-κB.
Противовоспалительная функция TRAF2 и TRAF3
Противовоспалительное действие
По сравнению с TRAF6, гораздо меньше известно о функции TRAF2 и TRAF3 в регулировании передачи сигналов TLR.Тем не менее, недавние исследования предполагают роль как TRAF2, так и TRAF3 в негативной регуляции TLR-стимулированной экспрессии провоспалительных цитокинов (9). Дефицит TRAF3 способствует индукции провоспалительных цитокинов и подавляет индукцию интерферона I типа в макрофагах (129, 130). Противовоспалительная функция TRAF3 была продемонстрирована на мышах с условным нокаутом TRAF3 (TRAF3-MKO) миелоидными клетками (126, 131). Специфичная для миелоидных клеток делеция TRAF3 не влияет на дифференцировку макрофагов, но делает мышей гиперчувствительными к индукции колита в модели сульфата натрия (DSS) (126).Мыши TRAF3-MKO также продуцируют повышенные уровни изотипов антител IgG3 и IgG2b в ответ на Т-независимые и Т-зависимые антигены соответственно, вероятно, из-за аберрантной продукции провоспалительных цитокинов, таких как IL-12 и IL-6 (131). . Соответственно, дефицит TRAF3 повышает чувствительность макрофагов к in vitro, индукция провоспалительных цитокинов с помощью лигандов TLR4 и TLR3, LPS и polyIC (126, 131). В более старшем возрасте (15–22 месяца) у мышей TRAF3-MKO спонтанно развивается хроническое воспаление во многих органах (131).Интересно, что у некоторых из этих мышей также развиваются опухоли, происходящие как из миелоидных клеток (гистиоцитов), так и из других типов клеток (включая В-клетки и гепатоциты), которые компетентны в экспрессии TRAF3 (131). Эти данные свидетельствуют о том, что хроническое воспаление у этих мутантных мышей может вносить вклад в онкогенез, хотя также возможно, что TRAF3 может регулировать опухолевую супрессивную функцию миелоидных клеток.
Неожиданным открытием является противовоспалительная функция TRAF2 в сигнальном пути TLR (126).Как и TRAF3, TRAF2 негативно регулирует индукцию провоспалительных цитокинов лигандами TLR, LPS и polyIC, а также цитокином IL-1β, а мыши с обусловленным миелоидными клетками нокаутом TRAF2 (TRAF2-MKO) гиперчувствительны к индукции колита в модели DSS. (126). Защитная роль TRAF2 при воспалении толстой кишки также была выявлена с использованием мышей с нокаутом по зародышевой линии TRAF2, у которых спонтанно развивается колит (132). Помимо своей противовоспалительной роли в миелоидных клетках, TRAF2, по-видимому, ингибирует воспаление, защищая эпителиальные клетки кишечника от апоптоза, индуцированного TNF (9, 132).На потенциальную роль TRAF2 и TRAF3 в регуляции воспалительного заболевания кишечника человека (IBD) указывает открытие, что уровень экспрессии этих членов TRAF повышается в ткани толстой кишки пациентов с IBD (133–135). Однако неясно, служит ли повышающая регуляция TRAF2 и TRAF3 механизмом обратной связи для подавления воспаления у людей.
Сигнальный механизм
Открытие того, что и TRAF2, и TRAF3 негативно регулируют индукцию провоспалительных цитокинов с помощью TLRs, поднимает вопрос о том, имеют ли эти члены TRAF общий механизм действия.Было показано, что TRAF3 играет роль в регуляции передачи сигналов, проксимальной к рецепторам в пути MyD88 (136) (рис. 3). В ответ на стимуляцию TLR как TRAF6, так и TRAF3 рекрутируются в сигнальный комплекс MyD88, в котором TRAF3, по-видимому, предотвращает перемещение комплекса TRAF6 с мембраны в цитоплазму, этап передачи сигнала, необходимый для активации нижележащих путей MAPK (136 ). Интересно, что передача сигналов TLR вызывает деградацию TRAF3, что важно для оптимальной активации MAPKs (136).Деградация TRAF3, по-видимому, включает TRAF6-опосредованное убиквитинирование K63 и активацию убиквитин-лигаз E3 cIAP1 и cIAP2, которые, в свою очередь, конъюгируют K48-связанные цепи убиквитина с TRAF3 и вызывают убиквитин-зависимую деградацию TRAF3 в протеасоме (136) (рис. 3). Распад TRAF3 также происходит в микроглии, резидентных макрофагах центральной нервной системы (ЦНС) (137). В микроглии TRAF3 подвергается деградации и ресинтезу наряду с индукцией экспериментального аутоиммунного энцефаломиелита (EAE), животной модели нейровоспалительного множественного склероза (137).Деградация TRAF3 также требует убиквитинлигазы E3 Peli1, которая обильно экспрессируется в микроглии. Дефицит Peli1 блокирует деградацию TRAF3 и вызывает его накопление, что способствует ослабленной индукции провоспалительных цитокинов и хемокинов в микроглии и улучшает патогенез ЕАЭ (137). Peli1, по-видимому, взаимодействует с TRAF6 или действует ниже TRAF6, чтобы опосредовать активацию cIAP, поскольку дефицит Peli1 ингибирует TRAF6-опосредованную индукцию убиквитинирования cIAP (137) (Рисунок 3).Роль TRAF3 в подавлении воспаления ЦНС также включает негативную регуляцию передачи сигналов IL-17R (138). TRAF3 связывается с IL-17R и препятствует образованию сигнального комплекса IL-17R-Act1-TRAF6 и стимулированной IL-17 активации путей NF-κB и MAPK. Ядерная Dbf2-родственная киназа 1 ингибирует связывание TRAF3 с IL-17R и, таким образом, способствует передаче сигналов IL-17R и воспалению (139).
Рисунок 3 . Противовоспалительная функция фактора, связанного с рецептором фактора некроза опухоли (TRAF) 2 и TRAF3 в сигнальном пути толл-подобного рецептора (TLR).В ответ на стимуляцию TLR TRAF6 рекрутируется в сигнальный комплекс MyD88. После активации комплекс TRAF6 перемещается с плазматической мембраны в цитоплазму, что является этапом передачи сигналов, необходимым для активации нижестоящего ядерного фактора κB и путей митоген-активируемой протеинкиназы (MAPK). TRAF3 негативно регулирует индуцированную TLR индукцию провоспалительных цитокинов посредством двух потенциальных механизмов. Первый направлен на нацеливание на комплекс MyD88-TRAF6 и предотвращает перемещение TRAF6 в цитоплазму, а второй — на совместную работу TRAF2 и cIAP, опосредуя убиквитин-зависимую деградацию двух основных провоспалительных факторов транскрипции, c-Rel и фактора регуляции интерферона. 5 (IRF5).Передача сигналов TLR временно отменяет функцию передачи отрицательных сигналов TRAF3, индуцируя протеолиз TRAF3. В этом процессе TRAF6 активирует cIAPs (cIAP1 и cIAP2) посредством убиквитинирования K63, которые, в свою очередь, действуют как лигазы убиквитина E3, опосредуя K48-связанное убиквитинирование и протеолиз TRAF3. В микроглии, Peli1, по-видимому, взаимодействует с TRAF6 или действует ниже TRAF6, опосредуя активацию cIAP.
Роль TRAF2 и TRAF3 в регуляции MAPK может зависеть от типа клеток, поскольку макрофаги, происходящие из костного мозга, и перитонеальные макрофаги, полученные от мышей TRAF2-MKO и TRAF3-MKO, не проявляют гипер-активацию MAPK при стимуляции LPS (126, 131 ).Другой потенциальный механизм, лежащий в основе противовоспалительной функции TRAF2 и TRAF3, — это подавление неканонической активации NF-κB. Как обсуждалось в вышеупомянутых разделах, TRAF2 и TRAF3 оба являются важными компонентами убиквитинлигазы TRAF-cIAP E3, которая обеспечивает деградацию NIK в неканоническом пути NF-κB (31). Дефицит TRAF2 или TRAF3 в макрофагах вызывает конститутивную активацию неканонического NF-κB, как показано накоплением NIK и процессингом p100 (126, 131). Однако неканоническая активация NF-κB, по-видимому, не играет важной роли, поскольку делеция NIK в макрофагах с дефицитом TRAF2 не предотвращает LPS-стимулированную гиперэкспрессию генов провоспалительных цитокинов, за исключением частичного ингибирования индукции Il23a . (126).По-видимому, TRAF2 и TRAF3 негативно регулируют два фактора транскрипции, c-Rel и фактор регуляции интерферона 5 (IRF5), которые принадлежат к семействам NF-κB и IRF, соответственно (126). IRF5 активируется TRAF6 в сигнальном пути MyD88, тогда как c-Rel активируется IKK посредством зависимой от фосфорилирования деградации ингибитора NF-κB IκBα (10, 18). И c-Rel, и IRF5 являются ключевыми медиаторами экспрессии провоспалительных цитокинов, стимулированной TLR (140–145). TRAF2 и TRAF3 контролируют устойчивый уровень экспрессии этих факторов транскрипции посттрансляционно, что включает механизм, зависимый от убиквитина и протеасомы (Рисунок 3).В макрофагах дикого типа c-Rel и IRF5 подвергаются конститутивной деградации, и для этого процесса требуются TRAF2 и TRAF3, поскольку делеция TRAF2 или TRAF3 вызывает стабилизацию и накопление c-Rel и IRF5 (126). Конститутивная деградация c-Rel и IRF5 также включает cIAP, поскольку инкубация макрофагов с маломолекулярным антагонистом IAP приводит к накоплению c-Rel и IRF5. Эти находки подтверждают возможность того, что убиквитинлигаза TRAF-cIAP E3 опосредует деградацию c-Rel и IRF5 в дополнение к NIK (Figure 3).
Новые данные свидетельствуют о том, что противовоспалительная функция комплекса TRAF – cIAP в макрофагах играет важную роль в регулировании противоопухолевого иммунитета. Специфичная для миелоидных клеток делеция TRAF2 способствует генерации макрофагов типа M1 в микроокружении опухоли, что способствует привлечению эффекторных Т-клеток CD4 и CD8 и повышает противоопухолевый иммунитет (126). Соответственно, как обсуждалось в предыдущем разделе, антагонисты IAP способствуют противоопухолевому иммунитету и действуют синергично с ингибиторами иммунных контрольных точек в мышиных моделях иммунотерапии рака (122–125).Эти иммуностимулирующие эффекты антагонистов IAP, вероятно, включают активацию как NIK-зависимого пути NF-κB, так и активацию макрофагов типа M1.
Заключительные замечания
Было установлено, что белки фактора, связанные с рецептором фактора некроза опухоли, в частности TRAF6 и TRAF2, являются основными медиаторами путей NF-κB и MAPK. Кроме того, недавние исследования идентифицировали TRAF2 и TRAF3 как негативные регуляторы неканонического пути NF-κB и провоспалительных путей передачи сигналов TLR.Накапливающиеся исследования также выявили потенциальные молекулярные механизмы, которые регулируют судьбу и сигнальные функции TRAFs. Эти исследования позволяют по-новому взглянуть на механизм, лежащий в основе сигнальной функции TRAF, и проливают свет на молекулярную основу заболеваний человека, связанных с TRAF-зависимыми сигнальными путями. Эти научные достижения также открыли новые возможности для разработки терапевтических подходов к лечению воспалительных заболеваний и рака.Например, антагонисты IAP, которые вызывают деградацию cIAP и, тем самым, нарушение комплекса TRAF – cIAP E3, были протестированы на животных моделях иммунотерапии рака и получили многообещающие результаты.
Недавние достижения также поднимают ряд вопросов, которые необходимо рассмотреть в будущих исследованиях. Например, молекулярный механизм, с помощью которого TRAF6 опосредует активацию путей NF-κB и MAPK, не полностью изучен. Хотя E3-лигазная активность TRAF6 важна, существуют разногласия относительно того, как TRAF6 реализует убиквитин-зависимый механизм активации путей NF-κB и MAPK.Сигнальный механизм TRAF2 также неуловим. В частности, обсуждается, требуется ли TRAF2 активность E3-лигазы для TNFα-индуцированной активации NF-κB. Более того, наши текущие знания в значительной степени основаны на исследованиях клеточных линий, и будет важно иметь модель на мышах для изучения роли убиквитинирования в функции TRAF2 в физиологических условиях. Другой важный вопрос касается отрицательной роли TRAFs в регуляции передачи сигналов. Важно дополнительно определить механизм, с помощью которого TRAF2 и TRAF3 функционируют вместе с cIAP, чтобы опосредовать убиквитин-зависимую деградацию белков и охарактеризовать целевые белки этого комплекса убиквитин-лигазы.Наконец, то, как белки TRAF регулируются в различных рецепторных путях, представляет собой еще одну важную область для будущих исследований.
Авторские взносы
Оба автора внесли значительный вклад в этот обзор.
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось в отсутствие каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Рецензенты HH и WZ и редактор, занимающийся обработкой, заявили о своей общей принадлежности.
Финансирование
Исследованияподдерживаются грантами Национальных институтов здравоохранения США (AI057555, AI064639, GM84459 и AI104519) для S-CS, а также грантами Национального фонда естественных наук Китая (№ 31301143) и Молодежного центра Хэбэй. Программа поддержки талантов для J-HS.
Сокращения
TNFR, рецептор фактора некроза опухоли; TRAF, фактор, связанный с рецептором фактора некроза опухоли; NF-κB, ядерный фактор κB; IκB, ингибирующий κB; IKK, IκB киназа; TAK1, трансформирующая бета-активированная киназа 1 фактора роста; NIK, киназа, индуцирующая NF-κB; BAFFR, рецептор BAFF; LTβR, рецептор β лимфотоксина; ERK, киназа, регулируемая внеклеточными сигналами; JNK, N-концевая киназа c-Jun; MAPK, митоген-активированная протеинкиназа; МКК, киназа МАРК; MAP3K, киназа киназы MAPK; PRR, рецептор распознавания образов; TLR, толл-подобный рецептор; NZF, Npl4 цинковый палец; IL-1R, рецептор IL-1; IRAK, киназа, связанная с IL-1R; RANK, активатор рецептора ядерного фактора каппа-B; MAVS, митохондриальный противовирусный сигнальный белок; MYSM1, myb-подобный SWIRM и домен 1 MPN; NLR, NOD-подобный рецептор; TRADD, домен смерти, связанный с рецептором TNF; RIP1, протеинкиназа 1, взаимодействующая с рецептором; S1P, сфингозин-1-фосфат; SphK1, сфингозинкиназа 1; DSS, сульфат натрия декстран; ВЗК, воспалительное заболевание кишечника; ЦНС, центральная нервная система; EAE, экспериментальный аутоиммунный энцефаломиелит; NDR1, ядерная киназа 1, родственная Dbf2; IRF5, фактор регуляции интерферона 5.
Список литературы
1. Ha H, Han D, Choi Y. TRAF-опосредованная передача сигналов TNFR-семейства. Curr Protoc Immunol (2009) Глава 11: Раздел 11.19D. DOI: 10.1002 / 0471142735.im1109ds87
CrossRef Полный текст | Google Scholar
4. Дэн Л., Ван С., Спенсер Э., Ян Л., Браун А., Ю Дж. И др. Активация комплекса киназы IkB с помощью TRAF6 требует димерного комплекса убиквитин-конъюгированного фермента и уникальной полиубиквитиновой цепи. Cell (2000) 103: 351–61.DOI: 10.1016 / S0092-8674 (00) 00126-4
CrossRef Полный текст | Google Scholar
6. Роте М., Вонг С.К., Хензель В.Дж., Геддел Д.В. Новое семейство предполагаемых сигнальных преобразователей, связанных с цитоплазматическим доменом рецептора фактора некроза опухоли 75 кДа. Cell (1994) 78: 681–92. DOI: 10.1016 / 0092-8674 (94)-0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8. Лю Т., Чжан Л., Джу Д., Сун К. Передача сигналов NF-kappaB при воспалении.